For more information about how the rapidtide library can be used, please see the API page. Common rapidtide workflows can also be called from the command line.
Background
Before talking about the individual programs, in the 2.0 release and going forward, I’ve tried to adhere to some common principals, across all program, to make them easier to understand and maintain, and more interoperable with other programs, and to simplify using the outputs.
BIDS Outputs
By default, all outputs are in BIDS compatible formats (this is most true for rapidtide and happy, which get the majority of the work, but the goal is to eventually make all the programs in the package conform to this). The two major ramifications of this are that I have tried to follow BIDS naming conventions for NIFTI, json, and text files containing time series. Also, all text files are by default BIDS continuous timeseries files - data is in compressed, tab separated column format (.tsv.gz), with the column names, sample rate, and start time, in the accompanying .json sidecar file.
Text Inputs
A side effect of moving to BIDS is that I’ve now made a standardized interface for reading text data into programs in the package to handle many different types of file. In general, now if you are asked for a timeseries, you can supply it in any of the following ways:
A plain text file with one or more columns.
You can specify any subset of columns in any order by adding “:colspec” to the end of the filename. “colspec” is a column specification consisting of one or more comma separated “column ranges”. A “column range” is either a single column number or a hyphen separated minimum and maximum column number. The first column in a file is column 0.
For example specifying, “mytextfile.txt:5-6,2,0,10-12”
would return an array containing all the timepoints from columns 5, 6, 2, 0, 10, 11, and 12 from mytextfile.txt, in that order. Not specifying “:colspec” returns all the columns in the file, in order.
If the program in question requires the actual sample rate, this can be specified
using the --samplerate or --sampletime flags. Otherwise 1.0Hz is assumed.
A BIDS continuous file with one or more columns.
BIDS files have names for each column, so these are used in column specification. For these files, “colspec” is a comma separated list of one or more column names:
“thefile_desc-interestingtimeseries_physio.json:cardiac,respiration”
would return the two named columns “cardiac” and “respiration” from the accompanying .tsv.gz file. Not specifying “:colspec” returns all the columns in the file, in order.
Because BIDS continuous files require sample rate and start time to be specified
in the sidecar file, these quantities will now already be set. Using the
--samplerate, --sampletime or --starttime flags will override any header
values, if specified.
Visualizing files
Any output NIFTI file can be visualized in your favorite NIFTI viewer. I like FSLeyes, part of FSL. It’s flexible and fast, and has lots of options for displaying 3 and 4D NIFTI files.
While there may be nice, general graphing tools for BIDS timeseries files, I wrote “showtc” many years ago, a matplotlib based file viewer with lots of nice tweaks to make pretty and informative graphs of various rapidtide input and output files. It’s part of rapidtide, and pretty easy to learn. Just type “showtc” with no arguments to get the options.
As an example, after running happy, if you want to see the derived cardiac waveform, you’d run:
showtc \
happytest_desc-slicerescardfromfmri_timeseries.json:cardiacfromfmri,cardiacfromfmri_dlfiltered \
--format separate
rapidtide
Description:
The central program in this package is rapidtide. This is the program that calculates a similarity function between a “probe” signal and every voxel of a BOLD fMRI dataset. It then determines the peak value, time delay, and wi dth of the similarity function to determine when and how strongly that probe signal appears in each voxel.
At its core, rapidtide is simply performing a full crosscorrelation between a “probe” timecourse and every voxel in an fMRI dataset (by “full” I mean over a range of time lags that account for any delays between the signals, rather than only at zero lag, as in a Pearson correlation). As with many things, however, the devil is in the details, and so rapidtide provides a number of features which make it pretty good at this particular task. A few highlights:
There are lots of ways to do something even as simple as a cross-correlation in a nonoptimal way (not windowing, improper normalization, doing it in the time rather than frequency domain, etc.). I’m pretty sure what rapidtide does by default is, if not the best way, at least a very good and very fast way.
rapidtide has been optimized and profiled to speed it up quite a bit; it has an optional dependency on numba – if it’s installed, some of the most heavily used routines will speed up significantly due to judicious use of @jit.
The sample rate of your probe regressor and the fMRI data do not have to match - rapidtide resamples the probe regressor to an integral multiple of the fMRI data rate automatically.
The probe and data can be temporally prefiltered to the LFO, respiratory, or cardiac frequency band with a command line switch, or you can specify any low, high, or bandpass range you want.
The data can be spatially smoothed at runtime (so you don’t have to keep smoothed versions of big datasets around). This is quite fast, so no reason not to do it this way.
rapidtide can generate a probe regressor from the global mean of the data itself - no externally recorded timecourse is required. Optionally you can input both a mask of regions that you want to be included in the mean, and the voxels that you want excluded from the mean (there are situations when you might want to do one or the other or both).
Determining the significance threshold for filtered correlations where the optimal delay has been selected is nontrivial; using the conventional formulae for the significance of a correlation leads to wildly inflated p values. rapidtide estimates the spurious correlation threshold by calculating the distribution of null correlation values obtained with a shuffling procedure at the beginning of each run (the default is to use 10000 shuffled correlations), and uses this value to mask the correlation maps it calculates. As of version 0.1.2 it will also handle two-tailed significance, which you need when using bipolar mode.
rapidtide can do an iterative refinement of the probe regressor by aligning the voxel timecourses in time and regenerating the test regressor.
rapidtide fits the peak of the correlation function, so you can make fine grained distinctions between close lag times. The resolution of the time lag discrimination is set by the length of the timecourse, not the timestep – this is a feature of correlations, not rapidtide.
Once the time delay in each voxel has been found, rapidtide outputs a 4D file of delayed probe regressors for using as voxel specific confound regressors or to estimate the strength of the probe regressor in each voxel. This regression is performed by default, but these outputs let you do it yourself if you are so inclined.
I’ve put a lot of effort into making the outputs as informative as possible - lots of useful maps, histograms, timecourses, etc.
There are a lot of tuning parameters you can mess with if you feel the need. I’ve tried to make intelligent defaults so things will work well out of the box, but you have the ability to set most of the interesting parameters yourself.
Inputs:
At a minimum, rapidtide needs a data file to work on (space by time), which is generally thought to be a BOLD fMRI data file. This can be Nifti1 or Nifti2 (for fMRI data, in which case it is time by up to 3 spatial dimensions) or a whitespace separated text file (for NIRS data, each column is a time course, each row a separate channel); I can currently read (probably) but not write Cifti files, so if you want to use grayordinate files you need to convert them to nifti2 in workbench, run rapidtide, then convert back. As soon as nibabel finishes their Cifti support (EDIT: and I get around to figuring it out), I’ll add that.
The file needs one time dimension and at least one spatial dimension. Internally, the array is flattened to a time by voxel array for simplicity.
The file you input here should be the result of any preprocessing you intend to do. The expectation is that rapidtide will be run as the last preprocessing step before resting state or task based analysis. So any slice time correction, motion correction, spike removal, etc. should already have been done. If you use FSL, this means that if you’ve run preprocessing, you would use the filtered_func_data.nii.gz file as input. Temporal and spatial filtering are the two (partial) exceptions here. Generally rapidtide is most useful for looking at low frequency oscillations, so when you run it, you usually use the --filterband lfo option or some other to limit the analysis to the detection and removal of low frequency systemic physiological oscillations. So rapidtide will generally apply it’s own temporal filtering on top of whatever you do in preprocessing. Also, you have the option of doing spatial smoothing in rapidtide to boost the SNR of the analysis; the hemodynamic signals rapidtide looks for are often very smooth, so you rather than smooth your functional data excessively, you can do it within rapidtide so that only the hemodynamic data is smoothed at that level.
Outputs:
Outputs are space or space by time NIFTI or text files, depending on what the input data file was, and some text files containing textual information, histograms, or numbers. File formats and naming follow BIDS conventions for derivative data for fMRI input data. Output spatial dimensions and file type match the input dimensions and file type (Nifti1 in, Nifti1 out). Depending on the file type of map, there can be no time dimension, a time dimension that matches the input file, or something else, such as a time lag dimension for a correlation map.
BIDS Outputs:
Name |
Extension(s) |
Content |
When present |
|---|---|---|---|
XXX_maxtime_map |
.nii.gz, .json |
Time of offset of the maximum of the similarity function |
Always |
XXX_desc-maxtime_hist |
.tsv, .json |
Histogram of the maxtime map |
Always |
XXX_maxcorr_map |
.nii.gz, .json |
Maximum similarity function value (usually the correlation coefficient, R) |
Always |
XXX_desc-maxcorr_hist |
.tsv, .json |
Histogram of the maxcorr map |
Always |
XXX_maxcorrsq_map |
.nii.gz, .json |
Maximum similarity function value, squared |
Always |
XXX_desc-maxcorrsq_hist |
.tsv, .json |
Histogram of the maxcorrsq map |
Always |
XXX_maxwidth_map |
.nii.gz, .json |
Width of the maximum of the similarity function |
Always |
XXX_desc-maxwidth_hist |
.tsv, .json |
Histogram of the maxwidth map |
Always |
XXX_MTT_map |
.nii.gz, .json |
Mean transit time (estimated) |
Always |
XXX_corrfit_mask |
.nii.gz |
Mask showing where the similarity function fit succeeded |
Always |
XXX_corrfitfailreason_map |
.nii.gz, .json |
A numerical code giving the reason a peak could not be found (0 if fit succeeded) |
Always |
XXX_desc-corrfitwindow_info |
.nii.gz |
Values used for correlation peak fitting |
Always |
XXX_desc-runoptions_info |
.json |
A detailed dump of all internal variables in the program. Useful for debugging and data provenance |
Always |
XXX_desc-lfofilterCleaned_bold |
.nii.gz, .json |
Filtered BOLD dataset after removing moving regressor |
If GLM filtering is enabled (default) |
XXX_desc-lfofilterRemoved_bold |
.nii.gz, .json |
Scaled, voxelwise delayed moving regressor that has been removed from the dataset |
If GLM filtering is enabled (default) and |
XXX_desc-lfofilterCoeff_map |
.nii.gz |
Magnitude of the delayed sLFO regressor from GLM filter |
If GLM filtering is enabled (default) |
XXX_desc-lfofilterMean_map |
.nii.gz |
Mean value over time, from GLM fit |
If GLM filtering is enabled (default) |
XXX_desc-lfofilterNorm_map |
.nii.gz |
GLM filter coefficient, divided by the voxel mean over time |
If GLM filtering is enabled (default) |
XXX_desc-lfofilterR_map |
.nii.gz |
R value for the GLM fit in the voxel |
If GLM filtering is enabled (default) |
XXX_desc-lfofilterR2_map |
.nii.gz |
R value for the GLM fit in the voxel, squared. Multiply by 100 to get percentage variance explained |
If GLM filtering is enabled (default) |
XXX_desc-CVR_map |
.nii.gz |
Cerebrovascular response, in units of % BOLD per unit of the supplied regressor (probably mmHg) |
If CVR mapping is enabled |
XXX_desc-CVRR_map |
.nii.gz |
R value for the CVR map fit in the voxel |
If CVR mapping is enabled |
XXX_desc-CVRR2_map |
.nii.gz |
R value for the CVR map fit in the voxel, squared. Multiply by 100 to get percentage variance explained |
If CVR mapping is enabled |
XXX_desc-processed_mask |
.nii.gz |
Mask of all voxels in which the similarity function is calculated |
Always |
XXX_desc-globalmean_mask |
.nii.gz |
Mask of voxels used to calculate the global mean signal |
This file will exist if no external regressor is specified |
XXX_desc-refine_mask |
.nii.gz |
Mask of voxels used in the last estimate a refined version of the probe regressor |
Present if passes > 1 |
XXX_desc-despeckle_mask |
.nii.gz |
Mask of the last set of voxels that had their time delays adjusted due to autocorrelations in the probe regressor |
Present if despecklepasses > 0 |
XXX_desc-corrout_info |
.nii.gz |
Full similarity function over the search range |
Always |
XXX_desc-gaussout_info |
.nii.gz |
Gaussian fit to similarity function peak over the search range |
Always |
XXX_desc-autocorr_timeseries |
.tsv, .json |
Autocorrelation of the probe regressor for each pass |
Always |
XXX_desc-corrdistdata_info |
.tsv, .json |
Null correlations from the significance estimation for each pass |
Present if |
XXX_desc-nullsimfunc_hist |
.tsv, .json |
Histogram of the distribution of null correlation values for each pass |
Present if |
XXX_desc-plt0p050_mask |
.nii.gz |
Voxels where the maxcorr value exceeds the p < 0.05 significance level |
Present if |
XXX_desc-plt0p010_mask |
.nii.gz |
Voxels where the maxcorr value exceeds the p < 0.01 significance level |
Present if |
XXX_desc-plt0p005_mask |
.nii.gz |
Voxels where the maxcorr value exceeds the p < 0.005 significance level |
Present if |
XXX_desc-plt0p001_mask |
.nii.gz |
Voxels where the maxcorr value exceeds the p < 0.001 significance level |
Present if |
XXX_desc-globallag_hist |
.tsv, .json |
Histogram of peak correlation times between probe and all voxels, over all time lags, for each pass |
Always |
XXX_desc-initialmovingregressor_timeseries |
.tsv, .json |
The raw and filtered initial probe regressor, at the original sampling resolution |
Always |
XXX_desc-movingregressor_timeseries |
.tsv, .json |
The probe regressor used in each pass, at the time resolution of the data |
Always |
XXX_desc-oversampledmovingregressor_timeseries |
.tsv, .json |
The probe regressor used in each pass, at the time resolution used for calculating the similarity function |
Always |
XXX_desc-refinedmovingregressor_timeseries |
.tsv, .json |
The raw and filtered probe regressor produced by the refinement procedure, at the time resolution of the data |
Present if passes > 1 |
Usage:
Perform a RIPTiDe time delay analysis on a dataset.
usage: rapidtide [-h] [--denoising | --delaymapping | --CVR]
[--venousrefine | --nirs]
[--datatstep TSTEP | --datafreq FREQ] [--noantialias]
[--invert] [--interptype {univariate,cubic,quadratic}]
[--offsettime OFFSETTIME] [--autosync]
[--filterband {None,vlf,lfo,resp,cardiac,hrv_ulf,hrv_vlf,hrv_lf,hrv_hf,hrv_vhf,lfo_legacy}]
[--filterfreqs LOWERPASS UPPERPASS]
[--filterstopfreqs LOWERSTOP UPPERSTOP]
[--filtertype {trapezoidal,brickwall,butterworth}]
[--butterorder ORDER] [--padseconds SECONDS]
[--permutationmethod {shuffle,phaserandom}] [--numnull NREPS]
[--skipsighistfit]
[--windowfunc {hamming,hann,blackmanharris,None}]
[--nowindow] [--zeropadding PADVAL] [--detrendorder ORDER]
[--spatialfilt GAUSSSIGMA] [--globalmean]
[--globalmaskmethod {mean,variance}] [--globalpreselect]
[--globalmeaninclude MASK[:VALSPEC]]
[--globalmeanexclude MASK[:VALSPEC]] [--motionfile MOTFILE]
[--motpos] [--motderiv] [--motdelayderiv]
[--globalsignalmethod {sum,meanscale,pca}]
[--globalpcacomponents VALUE] [--slicetimes FILE]
[--numskip SKIP] [--timerange START END] [--nothresh]
[--oversampfac OVERSAMPFAC] [--regressor FILE]
[--regressorfreq FREQ | --regressortstep TSTEP]
[--regressorstart START]
[--corrweighting {None,phat,liang,eckart,regressor}]
[--corrtype {linear,circular}]
[--corrmaskthresh PCT | --corrmask MASK[:VALSPEC]]
[--similaritymetric {correlation,mutualinfo,hybrid}]
[--mutualinfosmoothingtime TAU] [--simcalcrange START END]
[--fixdelay DELAYTIME | --searchrange LAGMIN LAGMAX]
[--sigmalimit SIGMALIMIT] [--bipolar] [--nofitfilt]
[--peakfittype {gauss,fastgauss,quad,fastquad,COM,None}]
[--despecklepasses PASSES] [--despecklethresh VAL]
[--refineprenorm {None,mean,var,std,invlag}]
[--refineweighting {None,NIRS,R,R2}] [--passes PASSES]
[--refineinclude MASK[:VALSPEC]]
[--refineexclude MASK[:VALSPEC]] [--norefinedespeckled]
[--lagminthresh MIN] [--lagmaxthresh MAX] [--ampthresh AMP]
[--sigmathresh SIGMA] [--offsetinclude MASK[:VALSPEC]]
[--offsetexclude MASK[:VALSPEC]] [--norefineoffset]
[--psdfilter] [--pickleft] [--pickleftthresh THRESH]
[--refineupperlag | --refinelowerlag]
[--refinetype {pca,ica,weighted_average,unweighted_average}]
[--pcacomponents VALUE] [--convergencethresh THRESH]
[--maxpasses MAXPASSES] [--nolimitoutput] [--savelags]
[--histlen HISTLEN] [--glmsourcefile FILE] [--noglm]
[--preservefiltering] [--saveintermediatemaps]
[--calccoherence] [--version] [--detailedversion]
[--noprogressbar] [--checkpoint] [--wiener] [--spcalculation]
[--dpoutput] [--cifti] [--simulate] [--displayplots]
[--nonumba] [--nosharedmem] [--memprofile]
[--mklthreads MKLTHREADS] [--nprocs NPROCS] [--reservecpu]
[--infotag tagname tagvalue]
[--corrbaselinespatialsigma SIGMA]
[--corrbaselinetemphpfcutoff FREQ]
[--spatialtolerance EPSILON] [--echocancel]
[--autorespdelete] [--noisetimecourse FILENAME[:VALSPEC]]
[--noisefreq FREQ | --noisetstep TSTEP] [--noisestart START]
[--noiseinvert] [--acfix] [--negativegradient]
[--cleanrefined] [--dispersioncalc] [--tmask FILE] [--debug]
[--verbose] [--disabledockermemfix] [--alwaysmultiproc]
[--singleproc_getNullDist] [--singleproc_calcsimilarity]
[--singleproc_peakeval] [--singleproc_fitcorr]
[--singleproc_refine] [--singleproc_makelaggedtcs]
[--singleproc_glm] [--isatest]
in_file outputname
Positional Arguments
- in_file
The input data file (BOLD fMRI file or NIRS text file).
- outputname
The root name for the output files. For BIDS compliance, this can only contain valid BIDS entities from the source data.
Analysis type
Single arguments that change default values for many arguments. Any parameter set by an analysis type can be overridden by setting that parameter explicitly. Analysis types are mutually exclusive with one another.
- --denoising
Preset for hemodynamic denoising - this is a macro that sets lagmin=-10.0, lagmax=10.0, passes=3, despeckle_passes=4, refineoffset=True, peakfittype=gauss, doglmfilt=True. Any of these options can be overridden with the appropriate additional arguments.
Default: False
- --delaymapping
Preset for delay mapping analysis - this is a macro that sets lagmin=-10.0, lagmax=30.0, passes=3, despeckle_passes=4, refineoffset=True, pickleft=True, limitoutput=True, doglmfilt=False. Any of these options can be overridden with the appropriate additional arguments.
Default: False
- --CVR
Preset for calibrated CVR mapping. Given an input regressor that represents some measured quantity over time (e.g. mmHg CO2 in the EtCO2 trace), rapidtide will calculate and output a map of percent BOLD change in units of the input regressor. To do this, this sets:passes=1, despeckle_passes=4, lagmin=-10.0, lagmax=30.0, and calculates a voxelwise GLM using the optimally delayed input regressor and the percent normalized, demeaned BOLD data as inputs. This map is output as (XXX_desc-CVR_map.nii.gz). If no input regressor is supplied, this will generate an error. These options can be overridden with the appropriate additional arguments.
Default: False
Macros
Single arguments that change default values for many arguments. Macros override individually set parameters. Macros are mutually exclusive with one another.
- --venousrefine
This is a macro that sets lagminthresh=2.5, lagmaxthresh=6.0, ampthresh=0.5, and refineupperlag to bias refinement towards voxels in the draining vasculature for an fMRI scan.
Default: False
- --nirs
This is a NIRS analysis - this is a macro that sets nothresh, refineprenorm=var, ampthresh=0.7, and lagminthresh=0.1.
Default: False
Preprocessing options
- --datatstep
Set the timestep of the data file to TSTEP. This will override the TR in an fMRI file. NOTE: if using data from a text file, for example with NIRS data, using one of these options is mandatory.
Default: auto
- --datafreq
Set the timestep of the data file to 1/FREQ. This will override the TR in an fMRI file. NOTE: if using data from a text file, for example with NIRS data, using one of these options is mandatory.
Default: auto
- --noantialias
Disable antialiasing filter.
Default: True
- --invert
Invert the sign of the regressor before processing.
Default: False
- --interptype
Possible choices: univariate, cubic, quadratic
Use specified interpolation type. Options are “cubic”, “quadratic”, and “univariate”. Default is univariate.
Default: “univariate”
- --offsettime
Apply offset OFFSETTIME to the lag regressors.
Default: 0.0
- --autosync
Estimate and apply the initial offsettime of an external regressor using the global crosscorrelation. Overrides offsettime if present.
Default: False
- --detrendorder
Set order of trend removal (0 to disable). Default is 3.
Default: 3
- --spatialfilt
Spatially filter fMRI data prior to analysis using GAUSSSIGMA in mm. Set GAUSSSIGMA negative to have rapidtide set it to half the mean voxel dimension (a rule of thumb for a good value).
Default: 0.0
- --globalmean
Generate a global mean regressor and use that as the reference regressor. If no external regressor is specified, this is enabled by default.
Default: False
- --globalmaskmethod
Possible choices: mean, variance
Select whether to use timecourse mean or variance to mask voxels prior to generating global mean. Default is “mean”.
Default: “mean”
- --globalpreselect
Treat this run as an initial pass to locate good candidate voxels for global mean regressor generation.
Default: False
- --globalmeaninclude
Only use voxels in mask file NAME for global regressor generation (if VALSPEC is given, only voxels with integral values listed in VALSPEC are used).
- --globalmeanexclude
Do not use voxels in mask file NAME for global regressor generation (if VALSPEC is given, only voxels with integral values listed in VALSPEC are excluded).
- --motionfile
Read 6 columns of motion regressors out of MOTFILE file (.par or BIDS .json) (with timepoints rows) and regress their derivatives and delayed derivatives out of the data prior to analysis.
- --motpos
Toggle whether displacement regressors will be used in motion regression. Default is False.
Default: False
- --motderiv
Toggle whether derivatives will be used in motion regression. Default is True.
Default: True
- --motdelayderiv
Toggle whether delayed derivative regressors will be used in motion regression. Default is False.
Default: False
- --globalsignalmethod
Possible choices: sum, meanscale, pca
The method for constructing the initial global signal regressor - straight summation, mean scaling each voxel prior to summation, or MLE PCA of the voxels in the global signal mask. Default is “sum.”
Default: “sum”
- --globalpcacomponents
Number of PCA components used for estimating the global signal. If VALUE >= 1, will retain thismany components. If 0.0 < VALUE < 1.0, enough components will be retained to explain the fraction VALUE of the total variance. If VALUE is negative, the number of components will be to retain will be selected automatically using the MLE method. Default is 0.8.
Default: 0.8
- --slicetimes
Apply offset times from FILE to each slice in the dataset.
- --numskip
SKIP TRs were previously deleted during preprocessing (e.g. if you have done your preprocessing in FSL and set dummypoints to a nonzero value.) Default is 0.
Default: 0
- --timerange
Limit analysis to data between timepoints START and END in the fmri file. If END is set to -1, analysis will go to the last timepoint. Negative values of START will be set to 0. Default is to use all timepoints.
Default: (-1, -1)
- --nothresh
Disable voxel intensity threshold (especially useful for NIRS data).
Default: False
Filtering options
- --filterband
Possible choices: None, vlf, lfo, resp, cardiac, hrv_ulf, hrv_vlf, hrv_lf, hrv_hf, hrv_vhf, lfo_legacy
Filter data and regressors to specific band. Use “None” to disable filtering. Default is “lfo”.
Default: “lfo”
- --filterfreqs
Filter data and regressors to retain LOWERPASS to UPPERPASS. If –filterstopfreqs is not also specified, LOWERSTOP and UPPERSTOP will be calculated automatically.
- --filterstopfreqs
Filter data and regressors to with stop frequencies LOWERSTOP and UPPERSTOP. LOWERSTOP must be <= LOWERPASS, UPPERSTOP must be >= UPPERPASS. Using this argument requires the use of –filterfreqs.
- --filtertype
Possible choices: trapezoidal, brickwall, butterworth
Filter data and regressors using a trapezoidal FFT, brickwall FFT, or butterworth bandpass filter. Default is “trapezoidal”.
Default: “trapezoidal”
- --butterorder
Set order of butterworth filter (if used). Default is 6.
Default: 6
- --padseconds
The number of seconds of padding to add to each end of a filtered timecourse to reduce end effects. Default is 30.0.
Default: 30.0
Significance calculation options
- --permutationmethod
Possible choices: shuffle, phaserandom
Permutation method for significance testing. Default is “shuffle”.
Default: “shuffle”
- --numnull
Estimate significance threshold by running NREPS null correlations (default is 10000, set to 0 to disable).
Default: 10000
- --skipsighistfit
Do not fit significance histogram with a Johnson SB function.
Default: True
Windowing options
- --windowfunc
Possible choices: hamming, hann, blackmanharris, None
Window function to use prior to correlation. Options are hamming, hann, blackmanharris, and None. Default is hamming
Default: “hamming”
- --nowindow
Disable precorrelation windowing.
Default: “hamming”
- --zeropadding
Pad input functions to correlation with PADVAL zeros on each side. A PADVAL of 0 does circular correlations, positive values reduce edge artifacts. Set PADVAL < 0 to set automatically. Default is 0.
Default: 0
Correlation options
- --oversampfac
Oversample the fMRI data by the following integral factor. Set to -1 for automatic selection (default).
Default: -1
- --regressor
Read the initial probe regressor from file FILE (if not specified, generate and use the global regressor).
- --regressorfreq
Probe regressor in file has sample frequency FREQ (default is 1/tr) NB: –regressorfreq and –regressortstep) are two ways to specify the same thing.
Default: auto
- --regressortstep
Probe regressor in file has sample frequency FREQ (default is 1/tr) NB: –regressorfreq and –regressortstep) are two ways to specify the same thing.
Default: auto
- --regressorstart
The time delay in seconds into the regressor file, corresponding in the first TR of the fMRI file (default is 0.0).
Default: 0.0
- --corrweighting
Possible choices: None, phat, liang, eckart, regressor
Method to use for cross-correlation weighting. Default is “None”.
Default: “None”
- --corrtype
Possible choices: linear, circular
Cross-correlation type (linear or circular). Default is “linear”.
Default: “linear”
- --corrmaskthresh
Do correlations in voxels where the mean exceeds this percentage of the robust max. Default is 1.0.
Default: 1.0
- --corrmask
Only do correlations in nonzero voxels in NAME (if VALSPEC is given, only voxels with integral values listed in VALSPEC are used).
- --similaritymetric
Possible choices: correlation, mutualinfo, hybrid
Similarity metric for finding delay values. Choices are “correlation”, “mutualinfo”, and “hybrid”. Default is correlation.
Default: “correlation”
- --mutualinfosmoothingtime
Time constant of a temporal smoothing function to apply to the mutual information function. Default is 3.0 seconds. TAU <=0.0 disables smoothing.
Default: 3.0
- --simcalcrange
Limit correlation caculation to data between timepoints START and END in the fmri file. If END is set to -1, analysis will go to the last timepoint. Negative values of START will be set to 0. Default is to use all timepoints.
Default: (-1, -1)
Correlation fitting options
- --fixdelay
Don’t fit the delay time - set it to DELAYTIME seconds for all voxels.
- --searchrange
Limit fit to a range of lags from LAGMIN to LAGMAX. Default is -30.0 to 30.0 seconds.
Default: (-30.0, 30.0)
- --sigmalimit
Reject lag fits with linewidth wider than SIGMALIMIT Hz. Default is 1000.0 Hz.
Default: 1000.0
- --bipolar
Bipolar mode - match peak correlation ignoring sign.
Default: False
- --nofitfilt
Do not zero out peak fit values if fit fails.
Default: True
- --peakfittype
Possible choices: gauss, fastgauss, quad, fastquad, COM, None
Method for fitting the peak of the similarity function “gauss” performs a Gaussian fit, and is most accurate. “quad” and “fastquad” use a quadratic fit, which is faster, but not as well tested. Default is “gauss”.
Default: “gauss”
- --despecklepasses
Detect and refit suspect correlations to disambiguate peak locations in PASSES passes. Default is to perform 4 passes. Set to 0 to disable.
Default: 4
- --despecklethresh
Refit correlation if median discontinuity magnitude exceeds VAL. Default is 5.0 seconds.
Default: 5.0
Regressor refinement options
- --refineprenorm
Possible choices: None, mean, var, std, invlag
Apply TYPE prenormalization to each timecourse prior to refinement. Default is “None”.
Default: “None”
- --refineweighting
Possible choices: None, NIRS, R, R2
Apply TYPE weighting to each timecourse prior to refinement. Default is “R2”.
Default: “R2”
- --passes
Set the number of processing passes to PASSES. Default is 3.
Default: 3
- --refineinclude
Only use voxels in file MASK for regressor refinement (if VALSPEC is given, only voxels with integral values listed in VALSPEC are used).
- --refineexclude
Do not use voxels in file MASK for regressor refinement (if VALSPEC is given, voxels with integral values listed in VALSPEC are excluded).
- --norefinedespeckled
Do not use despeckled pixels in calculating the refined regressor.
Default: True
- --lagminthresh
For refinement, exclude voxels with delays less than MIN. Default is 0.5 seconds.
Default: 0.5
- --lagmaxthresh
For refinement, exclude voxels with delays greater than MAX. Default is 5.0 seconds.
Default: 5.0
- --ampthresh
For refinement, exclude voxels with correlation coefficients less than AMP (default is 0.3). NOTE: ampthresh will automatically be set to the p<0.05 significance level determined by the –numnull option if NREPS is set greater than 0 and this is not manually specified.
Default: -1.0
- --sigmathresh
For refinement, exclude voxels with widths greater than SIGMA seconds. Default is 100.0 seconds.
Default: 100.0
- --offsetinclude
Only use voxels in file MASK for determining the zero time offset value (if VALSPEC is given, only voxels with integral values listed in VALSPEC are used).
- --offsetexclude
Do not use voxels in file MASK for determining the zero time offset value (if VALSPEC is given, voxels with integral values listed in VALSPEC are excluded).
- --norefineoffset
Disable realigning refined regressor to zero lag.
Default: True
- --psdfilter
Apply a PSD weighted Wiener filter to shifted timecourses prior to refinement.
Default: False
- --pickleft
Will select the leftmost delay peak when setting the refine offset.
Default: False
- --pickleftthresh
Threshhold value (fraction of maximum) in a histogram to be considered the start of a peak. Default is 0.33.
Default: 0.33
- --refineupperlag
Only use positive lags for regressor refinement.
Default: “both”
- --refinelowerlag
Only use negative lags for regressor refinement.
Default: “both”
- --refinetype
Possible choices: pca, ica, weighted_average, unweighted_average
Method with which to derive refined regressor. Default is “pca”.
Default: “pca”
- --pcacomponents
Number of PCA components used for refinement. If VALUE >= 1, will retain this many components. If 0.0 < VALUE < 1.0, enough components will be retained to explain the fraction VALUE of the total variance. If VALUE is negative, the number of components will be to retain will be selected automatically using the MLE method. Default is 0.8.
Default: 0.8
- --convergencethresh
Continue refinement until the MSE between regressors becomes <= THRESH. By default, this is not set, so refinement will run for the specified number of passes.
- --maxpasses
Terminate refinement after MAXPASSES passes, whether or not convergence has occured. Default is 15.
Default: 15
Output options
- --nolimitoutput
Save some of the large and rarely used files.
Default: True
- --savelags
Save a table of lagtimes used.
Default: False
- --histlen
Change the histogram length to HISTLEN. Default is 101.
Default: 101
- --glmsourcefile
Regress delayed regressors out of FILE instead of the initial fmri file used to estimate delays.
- --noglm
Turn off GLM filtering to remove delayed regressor from each voxel (disables output of fitNorm).
Default: True
- --preservefiltering
Don’t reread data prior to performing GLM.
Default: False
- --saveintermediatemaps
Save lag times, strengths, widths, and mask for each pass.
Default: False
- --calccoherence
Calculate and save the coherence between the final regressor and the data.
Default: False
Version options
- --version
show program’s version number and exit
- --detailedversion
show program’s version number and exit
Miscellaneous options
- --noprogressbar
Will disable showing progress bars (helpful if stdout is going to a file).
Default: True
- --checkpoint
Enable run checkpoints.
Default: False
- --wiener
Do Wiener deconvolution to find voxel transfer function.
Default: False
- --spcalculation
Use single precision for internal calculations (may be useful when RAM is limited).
Default: “double”
- --dpoutput
Use double precision for output files.
Default: “single”
- --cifti
Data file is a converted CIFTI.
Default: False
- --simulate
Simulate a run - just report command line options.
Default: False
- --displayplots
Display plots of interesting timecourses.
Default: False
- --nonumba
Disable jit compilation with numba.
Default: False
- --nosharedmem
Disable use of shared memory for large array storage.
Default: True
- --memprofile
Enable memory profiling - warning: this slows things down a lot.
Default: False
- --mklthreads
Use no more than MKLTHREADS worker threads in accelerated numpy calls.
Default: 1
- --nprocs
Use NPROCS worker processes for multiprocessing. Setting NPROCS to less than 1 sets the number of worker processes to n_cpus (unless –reservecpu is used).
Default: 1
- --reservecpu
When automatically setting nprocs, reserve one CPU for process management rather than using them all for worker threads.
Default: False
- --infotag
Additional key, value pairs to add to the options json file (useful for tracking analyses).
Experimental options (not fully tested, may not work)
- --corrbaselinespatialsigma
Spatial lowpass kernel, in mm, for filtering the correlation function baseline.
Default: 0.0
- --corrbaselinetemphpfcutoff
Temporal highpass cutoff, in Hz, for filtering the correlation function baseline.
Default: 0.0
- --spatialtolerance
When checking to see if the spatial dimensions of two NIFTI files match, allow a relative difference of EPSILON in any dimension. By default, this is set to 0.0, requiring an exact match.
Default: 0.0
- --echocancel
Attempt to perform echo cancellation.
Default: False
- --autorespdelete
Attempt to detect and remove respiratory signal that strays into the LFO band.
Default: False
- --noisetimecourse
Find and remove any instance of the timecourse supplied from any regressors used for analysis. (if VALSPEC is given, and there are multiple timecourses in the file, use the indicated timecourse.This can be the name of the regressor if it’s in the file, or the column number).
- --noisefreq
Noise timecourse in file has sample frequency FREQ (default is 1/tr) NB: –noisefreq and –noisetstep) are two ways to specify the same thing.
Default: auto
- --noisetstep
Noise timecourse in file has sample frequency FREQ (default is 1/tr) NB: –noisefreq and –noisetstep) are two ways to specify the same thing.
Default: auto
- --noisestart
The time delay in seconds into the noise timecourse file, corresponding in the first TR of the fMRI file (default is 0.0).
Default: 0.0
- --noiseinvert
Invert noise regressor prior to alignment.
Default: False
- --acfix
Check probe regressor for autocorrelations in order to disambiguate peak location.
Default: False
- --negativegradient
Calculate the negative gradient of the fmri data after spectral filtering so you can look for CSF flow à la https://www.biorxiv.org/content/10.1101/2021.03.29.437406v1.full.
Default: False
- --cleanrefined
Perform additional processing on refined regressor to remove spurious components.
Default: False
- --dispersioncalc
Generate extra data during refinement to allow calculation of dispersion.
Default: False
- --tmask
Only correlate during epochs specified in MASKFILE (NB: each line of FILE contains the time and duration of an epoch to include.
Debugging options. You probably don’t want to use any of these unless I ask you to to help diagnose a problem
- --debug
Enable additional debugging output.
Default: False
- --verbose
Enable additional runtime information output.
Default: False
- --disabledockermemfix
Disable docker memory limit setting.
Default: True
- --alwaysmultiproc
Use the multiprocessing code path even when nprocs=1.
Default: False
- --singleproc_getNullDist
Force single proc path for getNullDist.
Default: False
- --singleproc_calcsimilarity
Force single proc path for calcsimilarity.
Default: False
- --singleproc_peakeval
Force single proc path for peakeval.
Default: False
- --singleproc_fitcorr
Force single proc path for fitcorr.
Default: False
- --singleproc_refine
Force single proc path for refine.
Default: False
- --singleproc_makelaggedtcs
Force single proc path for makelaggedtcs.
Default: False
- --singleproc_glm
Force single proc path for glm.
Default: False
- --isatest
This run of rapidtide is in a unit test.
Default: False
Legacy interface:
For compatibility with old workflows, rapidtide can be called using legacy syntax by using “rapidtide2x_legacy”. Although the underlying code is the same, not all options are settable from the legacy interface. This interface is deprecated and will be removed in a future version of rapidtide, so please convert existing workflows.
usage: rapidtide2x_legacy datafilename outputname [-r LAGMIN,LAGMAX] [-s SIGMALIMIT] [-a] [--nowindow] [--phat] [--liang] [--eckart] [-f GAUSSSIGMA] [-O oversampfac] [-t TSTEP] [--datatstep=TSTEP] [--datafreq=FREQ] [-d] [-b] [-V] [-L] [-R] [-C] [-F LOWERFREQ,UPPERFREQ[,LOWERSTOP,UPPERSTOP]] [-o OFFSETTIME] [--autosync] [-T] [-p] [-P] [-B] [-h HISTLEN] [-i INTERPTYPE] [-I] [-Z DELAYTIME] [--nofitfilt] [--searchfrac=SEARCHFRAC] [-N NREPS] [--motionfile=MOTFILE] [--pickleft] [--numskip=SKIP] [--refineweighting=TYPE] [--refineprenorm=TYPE] [--passes=PASSES] [--refinepasses=PASSES] [--excluderefine=MASK] [--includerefine=MASK] [--includemean=MASK] [--excludemean=MASK][--lagminthresh=MIN] [--lagmaxthresh=MAX] [--ampthresh=AMP] [--sigmathresh=SIGMA] [--corrmask=MASK] [--corrmaskthresh=PCT] [--refineoffset] [--pca] [--ica] [--weightedavg] [--avg] [--psdfilter] [--noprogressbar] [--despecklethresh=VAL] [--despecklepasses=PASSES] [--dispersioncalc] [--refineupperlag] [--refinelowerlag] [--nosharedmem] [--tmask=MASKFILE] [--limitoutput] [--motionfile=FILENAME[:COLSPEC] [--softlimit] [--timerange=START,END] [--skipsighistfit] [--accheck] [--acfix][--numskip=SKIP] [--slicetimes=FILE] [--glmsourcefile=FILE] [--regressorfreq=FREQ] [--regressortstep=TSTEP][--regressor=FILENAME] [--regressorstart=STARTTIME] [--usesp] [--peakfittype=FITTYPE] [--mklthreads=NTHREADS] [--nprocs=NPROCS] [--nirs] [--venousrefine] Required arguments: datafilename - The input data file (BOLD fmri file or NIRS) outputname - The root name for the output files Optional arguments: Arguments are processed in order of appearance. Later options can override ones earlier on the command line Macros: --venousrefine - This is a macro that sets --lagminthresh=2.5, --lagmaxthresh=6.0, --ampthresh=0.5, and --refineupperlag to bias refinement towards voxels in the draining vasculature for an fMRI scan. --nirs - This is a NIRS analysis - this is a macro that sets --nothresh, --preservefiltering, --refinenorm=var, --ampthresh=0.7, and --lagminthresh=0.1. Preprocessing options: -t TSTEP, - Set the timestep of the data file to TSTEP (or 1/FREQ) --datatstep=TSTEP, This will override the TR in an fMRI file. --datafreq=FREQ NOTE: if using data from a text file, for example with NIRS data, using one of these options is mandatory. -a - Disable antialiasing filter --detrendorder=ORDER - Set order of trend removal (0 to disable, default is 1 - linear) -I - Invert the sign of the regressor before processing -i - Use specified interpolation type (options are 'cubic', 'quadratic', and 'univariate (default)') -o - Apply an offset OFFSETTIME to the lag regressors --autosync - Calculate and apply offset time of an external regressor from the global crosscorrelation. Overrides offsettime if specified. -b - Use butterworth filter for band splitting instead of trapezoidal FFT filter -F LOWERFREQ,UPPERFREQ[,LOWERSTOP,UPPERSTOP] - Filter data and regressors from LOWERFREQ to UPPERFREQ. LOWERSTOP and UPPERSTOP can be specified, or will be calculated automatically -V - Filter data and regressors to VLF band -L - Filter data and regressors to LFO band -R - Filter data and regressors to respiratory band -C - Filter data and regressors to cardiac band --padseconds=SECONDS - Set the filter pad time to SECONDS seconds. Default is 30.0 -N NREPS - Estimate significance threshold by running NREPS null correlations (default is 10000, set to 0 to disable). If you are running multiple passes, 'ampthresh' will be set to the 0.05 significance. level unless it is manually specified (see below). --permutationmethod=METHOD - Method for permuting the regressor for significance estimation. Default is shuffle --skipsighistfit - Do not fit significance histogram with a Johnson SB function --windowfunc=FUNC - Use FUNC window funcion prior to correlation. Options are hamming (default), hann, blackmanharris, and None --nowindow - Disable precorrelation windowing -f GAUSSSIGMA - Spatially filter fMRI data prior to analysis using GAUSSSIGMA in mm -M - Generate a global mean regressor and use that as the reference regressor --globalmeaninclude=MASK[:VALSPEC] - Only use voxels in NAME for global regressor generation (if VALSPEC is given, only voxels with integral values listed in VALSPEC are used.) --globalmeanexclude=MASK[:VALSPEC] - Do not use voxels in NAME for global regressor generation (if VALSPEC is given, only voxels with integral values listed in VALSPEC are used.) -m - Mean scale regressors during global mean estimation --slicetimes=FILE - Apply offset times from FILE to each slice in the dataset --numskip=SKIP - SKIP tr's were previously deleted during preprocessing (e.g. if you have done your preprocessing in FSL and set dummypoints to a nonzero value.) Default is 0. --timerange=START,END - Limit analysis to data between timepoints START and END in the fmri file. If END is set to -1, analysis will go to the last timepoint. Negative values of START will be set to 0. Default is to use all timepoints. --nothresh - Disable voxel intensity threshold (especially useful for NIRS data) --motionfile=MOTFILE[:COLSPEC] - Read 6 columns of motion regressors out of MOTFILE text file. (with timepoints rows) and regress their derivatives and delayed derivatives out of the data prior to analysis. If COLSPEC is present, use the comma separated list of ranges to specify X, Y, Z, RotX, RotY, and RotZ, in that order. For example, :3-5,7,0,9 would use columns 3, 4, 5, 7, 0 and 9 for X, Y, Z, RotX, RotY, RotZ, respectively --motpos - Toggle whether displacement regressors will be used in motion regression. Default is False. --motderiv - Toggle whether derivatives will be used in motion regression. Default is True. --motdelayderiv - Toggle whether delayed derivative regressors will be used in motion regression. Default is False. Correlation options: -O OVERSAMPFAC - Oversample the fMRI data by the following integral factor. Setting to -1 chooses the factor automatically (default) --regressor=FILENAME - Read probe regressor from file FILENAME (if none specified, generate and use global regressor) --regressorfreq=FREQ - Probe regressor in file has sample frequency FREQ (default is 1/tr) NB: --regressorfreq and --regressortstep are two ways to specify the same thing --regressortstep=TSTEP - Probe regressor in file has sample time step TSTEP (default is tr) NB: --regressorfreq and --regressortstep are two ways to specify the same thing --regressorstart=START - The time delay in seconds into the regressor file, corresponding in the first TR of the fmri file (default is 0.0) --phat - Use generalized cross-correlation with phase alignment transform (PHAT) instead of correlation --liang - Use generalized cross-correlation with Liang weighting function (Liang, et al, doi:10.1109/IMCCC.2015.283) --eckart - Use generalized cross-correlation with Eckart weighting function --corrmaskthresh=PCT - Do correlations in voxels where the mean exceeeds this percentage of the robust max (default is 1.0) --corrmask=MASK - Only do correlations in voxels in MASK (if set, corrmaskthresh is ignored). --accheck - Check for periodic components that corrupt the autocorrelation Correlation fitting options: -Z DELAYTIME - Don't fit the delay time - set it to DELAYTIME seconds for all voxels -r LAGMIN,LAGMAX - Limit fit to a range of lags from LAGMIN to LAGMAX -s SIGMALIMIT - Reject lag fits with linewidth wider than SIGMALIMIT -B - Bipolar mode - match peak correlation ignoring sign --nofitfilt - Do not zero out peak fit values if fit fails --searchfrac=FRAC - When peak fitting, include points with amplitude > FRAC * the maximum amplitude. (default value is 0.5) --peakfittype=FITTYPE - Method for fitting the peak of the similarity function (default is 'gauss'). 'quad' uses a quadratic fit. Other options are 'fastgauss' which is faster but not as well tested, and 'None'. --despecklepasses=PASSES - detect and refit suspect correlations to disambiguate peak locations in PASSES passes --despecklethresh=VAL - refit correlation if median discontinuity magnitude exceeds VAL (default is 5s) --softlimit - Allow peaks outside of range if the maximum correlation is at an edge of the range. Regressor refinement options: --refineprenorm=TYPE - Apply TYPE prenormalization to each timecourse prior to refinement (valid weightings are 'None', 'mean' (default), 'var', and 'std' --refineweighting=TYPE - Apply TYPE weighting to each timecourse prior to refinement (valid weightings are 'None', 'R', 'R2' (default) --passes=PASSES, - Set the number of processing passes to PASSES --refinepasses=PASSES (default is 1 pass - no refinement). NB: refinepasses is the wrong name for this option - --refinepasses is deprecated, use --passes from now on. --refineinclude=MASK[:VALSPEC] - Only use nonzero voxels in MASK for regressor refinement (if VALSPEC is given, only voxels with integral values listed in VALSPEC are used.) --refineexclude=MASK[:VALSPEC] - Do not use nonzero voxels in MASK for regressor refinement (if VALSPEC is given, only voxels with integral values listed in VALSPEC are used.) --lagminthresh=MIN - For refinement, exclude voxels with delays less than MIN (default is 0.5s) --lagmaxthresh=MAX - For refinement, exclude voxels with delays greater than MAX (default is 5s) --ampthresh=AMP - For refinement, exclude voxels with correlation coefficients less than AMP (default is 0.3). NOTE: ampthresh will automatically be set to the p<0.05 significance level determined by the -N option if -N is set greater than 0 and this is not manually specified. --sigmathresh=SIGMA - For refinement, exclude voxels with widths greater than SIGMA (default is 100s) --refineoffset - Adjust offset time during refinement to bring peak delay to zero --pickleft - When setting refineoffset, always select the leftmost histogram peak --pickleftthresh=THRESH - Set the threshold value (fraction of maximum) to decide something is a peak in a histogram. Default is 0.33. --refineupperlag - Only use positive lags for regressor refinement --refinelowerlag - Only use negative lags for regressor refinement --pca - Use pca to derive refined regressor (default is unweighted averaging) --ica - Use ica to derive refined regressor (default is unweighted averaging) --weightedavg - Use weighted average to derive refined regressor (default is unweighted averaging) --avg - Use unweighted average to derive refined regressor (default) --psdfilter - Apply a PSD weighted Wiener filter to shifted timecourses prior to refinement Output options: --limitoutput - Don't save some of the large and rarely used files -T - Save a table of lagtimes used -h HISTLEN - Change the histogram length to HISTLEN (default is 100) --glmsourcefile=FILE - Regress delayed regressors out of FILE instead of the initial fmri file used to estimate delays --noglm - Turn off GLM filtering to remove delayed regressor from each voxel (disables output of fitNorm) --preservefiltering - don't reread data prior to GLM Miscellaneous options: --noprogressbar - Disable progress bars - useful if saving output to files --wiener - Perform Wiener deconvolution to get voxel transfer functions --usesp - Use single precision for internal calculations (may be useful when RAM is limited) -c - Data file is a converted CIFTI -S - Simulate a run - just report command line options -d - Display plots of interesting timecourses --nonumba - Disable jit compilation with numba --nosharedmem - Disable use of shared memory for large array storage --memprofile - Enable memory profiling for debugging - warning: this slows things down a lot. --multiproc - Enable multiprocessing versions of key subroutines. This speeds things up dramatically. Almost certainly will NOT work on Windows (due to different forking behavior). --mklthreads=NTHREADS - Use no more than NTHREADS worker threads in accelerated numpy calls. --nprocs=NPROCS - Use NPROCS worker processes for multiprocessing. Setting NPROCS less than 1 sets the number of worker processes to n_cpus - 1 (default). Setting NPROCS enables --multiproc. --debug - Enable additional information output --saveoptionsasjson - Save the options file in json format rather than text. Will eventually become the default, but for now I'm just trying it out. Experimental options (not fully tested, may not work): --cleanrefined - perform additional processing on refined regressor to remove spurious components. --dispersioncalc - Generate extra data during refinement to allow calculation of dispersion. --acfix - Perform a secondary correlation to disambiguate peak location (enables --accheck). Experimental. --tmask=MASKFILE - Only correlate during epochs specified in MASKFILE (NB: if file has one colum, the length needs to match the number of TRs used. TRs with nonzero values will be used in analysis. If there are 2 or more columns, each line of MASKFILE contains the time (first column) and duration (second column) of an epoch to include.)
These options are somewhat self-explanatory. I will be expanding this section of the manual going forward, but I want to put something here to get this out here.
When using the legacy interface, file names will be output using the old, non-BIDS names and formats. rapidtide can be forced to use the old style outputs with the --legacyoutput flag.
Equivalence between BIDS and legacy outputs:
BIDS style name |
Legacy name |
|---|---|
XXX_maxtime_map(.nii.gz, .json) |
XXX_lagtimes.nii.gz |
XXX_desc-maxtime_hist(.tsv, .json) |
XXX_laghist.txt |
XXX_maxcorr_map(.nii.gz, .json) |
XXX_lagstrengths.nii.gz |
XXX_desc-maxcorr_hist(.tsv, .json) |
XXX_strengthhist.txt |
XXX_maxcorrsq_map(.nii.gz, .json) |
XXX_R2.nii.gz |
XXX_desc-maxcorrsq_hist(.tsv, .json) |
XXX_R2hist.txt |
XXX_maxwidth_map(.nii.gz, .json) |
XXX_lagsigma.nii.gz |
XXX_desc-maxwidth_hist(.tsv, .json) |
XXX_widthhist.txt |
XXX_MTT_map(.nii.gz, .json) |
XXX_MTT.nii.gz |
XXX_corrfit_mask.nii.gz |
XXX_fitmask.nii.gz |
XXX_corrfitfailreason_map(.nii.gz, .json) |
XXX_failreason.nii.gz |
XXX_desc-corrfitwindow_info.nii.gz |
XXX_windowout.nii.gz |
XXX_desc-runoptions_info.json |
XXX_options.json |
XXX_desc-lfofilterCleaned_bold(.nii.gz, .json) |
XXX_filtereddata.nii.gz |
XXX_desc-lfofilterRemoved_bold(.nii.gz, .json) |
XXX_datatoremove.nii.gz |
XXX_desc-lfofilterCoeff_map.nii.gz |
XXX_fitcoeff.nii.gz |
XXX_desc-lfofilterMean_map.nii.gz |
XXX_meanvalue.nii.gz |
XXX_desc-lfofilterNorm_map.nii.gz |
XXX_fitNorm.nii.gz |
XXX_desc-lfofilterR2_map.nii.gz |
XXX_r2value.nii.gz |
XXX_desc-lfofilterR_map.nii.gz |
XXX_rvalue.nii.gz |
XXX_desc-processed_mask.nii.gz |
XXX_corrmask.nii.gz |
XXX_desc-globalmean_mask.nii.gz |
XXX_meanmask.nii.gz |
XXX_desc-refine_mask.nii.gz |
XXX_refinemask.nii.gz |
XXX_desc-despeckle_mask.nii.gz |
XXX_despecklemask.nii.gz |
XXX_desc-corrout_info.nii.gz |
XXX_corrout.nii.gz |
XXX_desc-gaussout_info.nii.gz |
XXX_gaussout.nii.gz |
XXX_desc-autocorr_timeseries(.tsv, .json) |
XXX_referenceautocorr_passN.txt |
XXX_desc-corrdistdata_info(.tsv, .json) |
XXX_corrdistdata_passN.txt |
XXX_desc-nullsimfunc_hist(.tsv, .json) |
XXX_nullsimfunchist_passN.txt |
XXX_desc-plt0p050_mask.nii.gz |
XXX_p_lt_0p050_mask.nii.gz |
XXX_desc-plt0p010_mask.nii.gz |
XXX_p_lt_0p010_mask.nii.gz |
XXX_desc-plt0p005_mask.nii.gz |
XXX_p_lt_0p005_mask.nii.gz |
XXX_desc-plt0p001_mask.nii.gz |
XXX_p_lt_0p001_mask.nii.gz |
XXX_desc-globallag_hist(.tsv, .json) |
XXX_globallaghist_passN.txt |
XXX_desc-initialmovingregressor_timeseries(.tsv, .json) |
XXX_reference_origres.txt, XXX_reference_origres_prefilt.txt |
XXX_desc-movingregressor_timeseries(.tsv, .json) |
XXX_reference_fmrires_passN.txt |
XXX_desc-oversampledmovingregressor_timeseries(.tsv, .json) |
XXX_reference_resampres_passN.txt |
XXX_desc-refinedmovingregressor_timeseries(.tsv, .json) |
XXX_unfilteredrefinedregressor_passN.txt, XXX_refinedregressor_passN.txt |
XXX_commandline.txt |
XXX_commandline.txt |
XXX_formattedcommandline.txt |
XXX_formattedcommandline.txt |
XXX_memusage.csv |
XXX_memusage.csv |
XXX_runtimings.txt |
XXX_runtimings.txt |
Examples:
Rapidtide can do many things - as I’ve found more interesting things to do with time delay processing, it’s gained new functions and options to support these new applications. As a result, it can be a little hard to know what to use for a new experiment. To help with that, I’ve decided to add this section to the manual to get you started. It’s broken up by type of data/analysis you might want to do.
Removing low frequency physiological noise from resting state data
This is what I thought most people would use rapidtide for - finding and removing the low frequency (LFO) signal from an existing dataset. This presupposes you have not made a simultaneous physiological recording (well, you may have, but it assumes you aren’t using it). For this, you can use a minimal set of options, since the defaults are mostly right.
The base command you’d use would be:
rapidtide inputfmrifile outputname --frequencyband lfo --passes 3
This will do a fairly simple analysis. First, the -L option means that rapidtide will prefilter the data to the LFO band (0.009-0.15Hz). It will then construct a regressor from the global mean of the signal in inputfmrifile (default behavior if no regressor is specified), and then use crosscorrelation to determine the time delay in each voxel. The --passes=3 option directs rapidtide to to perform the delay analysis 3 times, each time generating a new estimate of the global noise signal by aligning all of the timecourses in the data to bring the global signal in phase prior to averaging. The --refineoffset flag recenters the peak of the delay distribution on zero during the refinement process, which should make datasets easier to compare. After the three passes are complete, it will then use a GLM filter to remove a lagged copy of the final mean regressor that from the data - this denoised data will be in the file “outputname_filtereddata.nii.gz”. There will also a number of maps output with the prefix “outputname_” of delay, correlation strength and so on.
Mapping long time delays in response to a gas challenge experiment:
Processing this sort of data requires a very different set of options from the previous case. Instead of the distribution of delays you expect in healthy controls (a slightly skewed, somewhat normal distribution with a tail on the positive side, ranging from about -5 to 5 seconds), in this case, the maximum delay can be extremely long (100-120 seconds is not uncommon in stroke, moyamoya disesase, and atherosclerosis). To do this, you need to radically change what options you use, not just the delay range, but a number of other options having to do with refinement and statistical measures.
For this type of analysis, a good place to start is the following:
rapidtide inputfmrifile outputname --numnull 0 --searchrange -10 140 --filterfreqs 0.0 0.1 --ampthresh 0.2 --noglm --nofitfilt
The first option (--numnull 0), shuts off the calculation of the null correlation distribution. This is used to determine the significance threshold, but the method currently implemented in rapidtide is a bit simplistic - it assumes that all the time points in the data are exchangable. This is certainly true for resting state data (see above), but it is very much NOT true for block paradigm gas challenges. To properly analyze those, I need to consider what time points are ‘equivalent’, and up to now, I don’t, so setting the number of iterations in the Monte Carlo analysis to zero omits this step.
The second option (--searchrange -10 140) is fairly obvious - this extends the detectable delay range out to 140 seconds. Note that this is somewhat larger than the maximum delays we frequently see, but to find the correlation peak with maximum precision, you need sufficient additional delay values so that the correlation can come to a peak and then come down enough that you can properly fit it.
Setting --filterfreqs 0.0 0.1 is VERY important. By default, rapidtide assumes you are looking at endogenous low frequency oscillations, which typically between 0.09 and 0.15 Hz. However, gas challenge paradigms are usually MUCH lower frequency (90 seconds off, 90 seconds on corresponds to 1/180s = ~0.006Hz). So if you use the default frequency settings, you will completely filter out your stimulus, and presumably, your response. If you are processing one of these experiments and get no results whatsoever, this is almost certainly the problem.
The --noglm option disables data filtering. If you are using rapidtide to estimate and remove low frequency noise from resting state or task fMRI data, the last step is to use a glm filter to remove this circulatory signal, leaving “pure” neuronal signal, which you’ll use in further analyses. That’s not relevant here - the signal you’d be removing is the one you care about. So this option skips that step to save time and disk space.
--nofitfilt skips a step after peak estimation. Estimating the delay and correlation amplitude in each voxel is a two step process. First you make a quick estimate (where is the maximum point of the correlation function, and what is its amplitude?), then you refine it by fitting a Gaussian function to the peak to improve the estimate. If this step fails, which it can if the peak is too close to the end of the lag range, or strangely shaped, the default behavior is to mark the point as bad and zero out the parameters for the voxel. The nofitfilt option means that if the fit fails, output the initial estimates rather than all zeros. This means that you get some information, even if it’s not fully refined. In my experience it does tend to make the maps for the gas challenge experiments a lot cleaner to use this option since the correlation function is pretty well behaved.
CVR mapping:
This is a slightly different twist on interpreting the strength of the lagged correlation. In this case, you supply an input regressor that corresponds to a measured, calibrated CO2 quantity (for example, etCO2 in mmHg). Rapidtide then does a modified analysis - it still uses the cross-correlation to find when the input regressor is maximally aligned with the variance in the voxel signal, but instead of only returning a correlation strength, it calculates the percentage BOLD change in each voxel in units of the input regressor (e.g. %BOLD/mmHg), which is the standard in CVR analysis.
You invoke this with the –CVR option.
To do this, I disabled refinement, hijacked the GLM filtering routine, and messed with some normalizations. If you want to refine your regressor estimate, or filter the sLFO signal out of your data, you need to do a separate analysis.
Denoising NIRS data:
When we started this whole research effort, I waw originally planning to denoise NIRS data, not fMRI data. But one thing led to another, and the NIRS got derailed for the fMRI effort. Now that we have some time to catch our breaths, and more importantly, we have access to some much higher quality NIRS data, this moved back to the front burner. The majority of the work was already done, I just needed to account for a few qualities that make NIRS data different from fMRI data:
NIRS data is not generally stored in NIFTI files. There is not as yet a standard NIRS format. In the absence of one, you could do worse than a multicolumn text file, with one column per data channel. That’s what I did here - if the file has a ‘.txt’ extension rather than ‘.nii.’, ‘.nii.gz’, or no extension, it will assume all I/O should be done on multicolumn text files.
NIRS data is often zero mean. This turned out to mess with a lot of my assumptions about which voxels have significant data, and mask construction. This has led to some new options for specifying mask threshholds and data averaging.
NIRS data is in some sense “calibrated” as relative micromolar changes in oxy-, deoxy-, and total hemoglobin concentration, so mean and/or variance normalizing the timecourses may not be right thing to do. I’ve added in some new options to mess with normalizations.
happy
Description:
happy is a new addition to the rapidtide suite. It’s complementary to rapidtide - it’s focussed on fast, cardiac signals in fMRI, rather than the slow, LFO signals we are usually looking at. It’s sort of a Frankenprogram - it has three distinct jobs, which are related, but are very distinct.
The first thing happy does is try to extract a cardiac waveform from the fMRI data. This is something I’ve been thinking about for a long time. Words go here
The second task is to take this raw estimate of the cardiac waveform, and clean it up using a deep learning filter. The original signal is useful, but pretty gross, but I figured you should be able to exploit the pseudoperiodic nature of the signal to greatly improve it. This is also a testbed to work on using neural nets to process time domain signals. It seemed like a worthwhile project, so it got grafted in.
The final task (which was actually the initial task, and the reason I wrote happy to begin with) is to implement Henning Voss’ totally cool hypersampling with analytic phase projection (guess where the name “happy” comes from). This is fairly straightforward, as Voss describes his method very clearly. But I have lots of data with no simultaneously recorded cardiac signals, and I was too lazy to go find datasets with pleth data to play with, so that’s why I did the cardiac waveform extraction part.
Inputs:
Happy needs a 4D BOLD fMRI data file (space by time) as input. This can be Nifti1 or Nifti2. If you have a simultaneously recorded cardiac waveform, it will happily use it, otherwise it will try to construct (and refine) one. NOTE: the 4D input dataset needs to be completely unpreprocessed - gradient distortion correction and motion correction can destroy the relationship between slice number and actual acquisition time, and slice time correction does not behave as expected for aliased signals (which the cardiac component in fMRI most certainly is), and in any case we need the slice time offsets to construct our waveform.
Outputs:
Outputs are space or space by time Nifti or text files, depending on what the input data file was, and some text files containing textual information, histograms, or numbers. File formats and naming follow BIDS conventions for derivative data for fMRI input data. Output spatial dimensions and file type match the input dimensions and file type (Nifti1 in, Nifti1 out). Depending on the file type of map, there can be no time dimension, a time dimension that matches the input file, or something else, such as a time lag dimension for a correlation map.
BIDS Outputs:
Name |
Extension(s) |
Content |
When present |
|---|---|---|---|
XXX_commandline |
.txt |
The command line used to run happy |
Always |
XXX_formattedcommandline |
.txt |
The command line used to run happy, attractively formatted |
Always |
XXX_desc-rawapp_info |
.nii.gz |
The analytic phase projection map of the cardiac waveform |
Always |
XXX_desc-app_info |
.nii.gz |
The analytic phase projection map of the cardiac waveform, voxelwise minimum subtracted |
Always |
XXX_desc-normapp_info |
.nii.gz |
The analytic phase projection map of the cardiac waveform, voxelwise minimum subtracted and normalized |
Always |
XXX_desc-apppeaks_hist |
.tsv.gz, .json |
Not sure |
Always |
XXX_desc-apppeaks_hist_centerofmass |
.txt |
Not sure |
Always |
XXX_desc-apppeaks_hist_peak |
.txt |
Not sure |
Always |
XXX_desc-slicerescardfromfmri_timeseries |
.tsv.gz, .json |
Cardiac timeseries at the time resolution of slice acquisition ((1/TR * number of slices / multiband factor |
Always |
XXX_desc-stdrescardfromfmri_timeseries |
.tsv.gz, .json |
Cardiac timeseries at standard time resolution (25.O Hz) |
Always |
XXX_desc-cardpulsefromfmri_timeseries |
.tsv.gz, .json |
The average (over time from minimum) of the cardiac waveform over all voxels |
Always |
XXX_desc-cardiaccyclefromfmri_timeseries |
.tsv.gz, .json |
The average (over a single cardiac cycle) of the cardiac waveform over all voxels |
Always |
XXX_desc-cine_info |
.nii.gz |
Average image of the fMRI data over a single cardiac cycle |
Always |
XXX_desc-cycleaverage_timeseries |
.tsv.gz, .json |
Not sure |
Always |
XXX_desc-maxphase_map |
.nii.gz |
Map of the average phase where the maximum amplitude occurs for each voxel |
Always |
XXX_desc-minphase_map |
.nii.gz |
Map of the average phase where the minimum amplitude occurs for each voxel |
Always |
XXX_desc-processvoxels_mask |
.nii.gz |
Map of all voxels used for analytic phase projection |
Always |
XXX_desc-vessels_map |
.nii.gz |
Amplitude of variance over a cardiac cycle (large values are assumed to be vessels) |
Always |
XXX_desc-vessels_mask |
.nii.gz |
Locations of voxels with variance over a cardiac cycle that exceeds a threshold (assumed to be vessels) |
Always |
XXX_desc-arteries_map |
.nii.gz |
High variance vessels with early maximum values within the cardiac cycle |
Always |
XXX_desc-veins_map |
.nii.gz |
High variance vessels with late maximum values within the cardiac cycle |
Always |
XXX_info |
.json |
Run parameters and derived values found during the run (quality metrics, derived thresholds, etc.) |
Always |
XXX_memusage |
.csv |
Memory statistics at multiple checkpoints over the course of the run |
Always |
XXX_runtimings |
.txt |
Detailed timing information |
Always |
Usage:
Hypersampling by Analytic Phase Projection - Yay!.
usage: happy [-h] [--cardcalconly] [--skipdlfilter]
[--usesuperdangerousworkaround] [--slicetimesareinseconds]
[--model MODELNAME] [--mklthreads NTHREADS] [--numskip SKIP]
[--motskip SKIP] [--motionfile MOTFILE] [--motionhp HPFREQ]
[--motionlp LPFREQ] [--nomotorthogonalize] [--motpos]
[--nomotderiv] [--nomotderivdelayed] [--discardmotionfiltered]
[--estmask MASKNAME] [--minhr MINHR] [--maxhr MAXHR]
[--minhrfilt MINHR] [--maxhrfilt MAXHR]
[--hilbertcomponents NCOMPS] [--envcutoff CUTOFF]
[--notchwidth WIDTH] [--invertphysiosign]
[--cardiacfile FILE[:COL]]
[--cardiacfreq FREQ | --cardiactstep TSTEP]
[--cardiacstart START] [--forcehr BPM]
[--respirationfile FILE[:COL]]
[--respirationfreq FREQ | --respirationtstep TSTEP]
[--respirationstart START] [--forcerr BreathsPM] [--spatialglm]
[--temporalglm] [--stdfreq FREQ] [--outputbins BINS]
[--gridbins BINS] [--gridkernel {old,gauss,kaiser}]
[--projmask MASKNAME] [--projectwithraw] [--fliparteries]
[--arteriesonly] [--version] [--detailedversion]
[--aliasedcorrelation] [--upsample] [--estimateflow]
[--noprogressbar] [--infotag tagname tagvalue] [--debug]
[--nodetrend DETRENDORDER] [--noorthog] [--disablenotch]
[--nomask] [--nocensor] [--noappsmooth] [--nophasefilt]
[--nocardiacalign] [--saveinfoastext] [--saveintermediate]
[--increaseoutputlevel] [--decreaseoutputlevel]
fmrifilename slicetimename outputroot
Positional Arguments
- fmrifilename
The input data file (BOLD fmri file or NIRS text file)
- slicetimename
Text file containing the offset time in seconds of each slice relative to the start of the TR, one value per line, OR the BIDS sidecar JSON file.NB: FSL slicetime files give slice times in fractions of a TR, BIDS sidecars give slice times in seconds. Non-json files are assumed to be the FSL style (fractions of a TR) UNLESS the –slicetimesareinseconds flag is used.
- outputroot
The root name for the output files
Processing steps
- --cardcalconly
Stop after all cardiac regressor calculation steps (before phase projection).
Default: False
- --skipdlfilter
Disable deep learning cardiac waveform filter.
Default: True
- --usesuperdangerousworkaround
Some versions of tensorflow seem to have some weird conflict with MKL whichI don’t seem to be able to fix. If the dl filter bombs complaining about multiple openmp libraries, try rerunning with the secret and inadvisable ‘–usesuperdangerousworkaround’ flag. Good luck!
Default: False
- --slicetimesareinseconds
If a non-json slicetime file is specified, happy assumes the file is FSL style (slice times are specified in fractions of a TR). Setting this flag overrides this assumption, and interprets the slice time file as being in seconds. This does nothing when the slicetime file is a .json BIDS sidecar.
Default: False
- --model
Use model MODELNAME for dl filter (default is model_revised - from the revised NeuroImage paper.
Default: “model_revised”
Performance
- --mklthreads
Use NTHREADS MKL threads to accelerate processing (defaults to 1 - more threads up to the number of cores can accelerate processing a lot, but can really kill you on clusters unless you’re very careful. Use at your own risk
Default: 1
Preprocessing
- --numskip
Skip SKIP tr’s at the beginning of the fMRI file (default is 0).
Default: 0
- --motskip
Skip SKIP tr’s at the beginning of the motion regressor file (default is 0).
Default: 0
- --motionfile
Read 6 columns of motion regressors out of MOTFILE file (.par or BIDS .json) (with timepoints rows) and regress them, their derivatives, and delayed derivatives out of the data prior to analysis.
- --motionhp
Highpass filter motion regressors to HPFREQ Hz prior to regression.
- --motionlp
Lowpass filter motion regressors to LPFREQ Hz prior to regression.
- --nomotorthogonalize
Do not orthogonalize motion regressors prior to regressing them out of the data.
Default: True
- --motpos
Include motion position regressors.
Default: False
- --nomotderiv
Do not use motion derivative regressors.
Default: True
- --nomotderivdelayed
Do not use delayed motion derivative regressors.
Default: True
- --discardmotionfiltered
Do not save data after motion filtering.
Default: True
Cardiac estimation tuning
- --estmask
Generation of cardiac waveform from data will be restricted to voxels in MASKNAME and weighted by the mask intensity. If this is selected, happy will only make a single pass through the data (the initial vessel mask generation pass will be skipped).
- --minhr
Limit lower cardiac frequency search range to MINHR BPM (default is 40).
Default: 40.0
- --maxhr
Limit upper cardiac frequency search range to MAXHR BPM (default is 140).
Default: 140.0
- --minhrfilt
Highpass filter cardiac waveform estimate to MINHR BPM (default is 40).
Default: 40.0
- --maxhrfilt
Lowpass filter cardiac waveform estimate to MAXHR BPM (default is 1000).
Default: 1000.0
- --hilbertcomponents
Retain NCOMPS components of the cardiac frequency signal to Hilbert transform (default is 1).
Default: 1
- --envcutoff
Lowpass filter cardiac normalization envelope to CUTOFF Hz (default is 0.4 Hz).
Default: 0.4
- --notchwidth
Set the width of the notch filter, in percent of the notch frequency (default is 1.5).
Default: 1.5
- --invertphysiosign
Invert the waveform extracted from the physiological signal. Use this if there is a contrast agent in the blood.
Default: False
External cardiac waveform options
- --cardiacfile
Read the cardiac waveform from file FILE. If COL is an integer, and FILE is a text file, use the COL’th column. If FILE is a BIDS format json file, use column named COL. If no file is specified, estimate the cardiac signal from the fMRI data.
- --cardiacfreq
Cardiac waveform in cardiacfile has sample frequency FREQ (default is 32Hz). NB: –cardiacfreq and –cardiactstep are two ways to specify the same thing.
Default: -32.0
- --cardiactstep
Cardiac waveform in cardiacfile has time step TSTEP (default is 1/32 sec). NB: –cardiacfreq and –cardiactstep are two ways to specify the same thing.
Default: -32.0
- --cardiacstart
The time delay in seconds into the cardiac file, corresponding to the first TR of the fMRI file (default is 0.0)
- --forcehr
Force heart rate fundamental detector to be centered at BPM (overrides peak frequencies found from spectrum). Usefulif there is structured noise that confuses the peak finder.
External respiration waveform options
- --respirationfile
Read the respiration waveform from file FILE. If COL is an integer, and FILE is a text file, use the COL’th column. If FILE is a BIDS format json file, use column named COL.
- --respirationfreq
Respiration waveform in respirationfile has sample frequency FREQ (default is 32Hz). NB: –respirationfreq and –respirationtstep are two ways to specify the same thing.
Default: -32.0
- --respirationtstep
Respiration waveform in respirationfile has time step TSTEP (default is 1/32 sec). NB: –respirationfreq and –respirationtstep are two ways to specify the same thing.
Default: -32.0
- --respirationstart
The time delay in seconds into the respiration file, corresponding to the first TR of the fMRI file (default is 0.0)
- --forcerr
Force respiratory rate fundamental detector to be centered at BreathsPM (overrides peak frequencies found from spectrum). Usefulif there is structured noise that confuses the peak finder.
Output processing
- --spatialglm
Generate framewise cardiac signal maps and filter them out of the input data.
Default: False
- --temporalglm
Generate voxelwise aliased synthetic cardiac regressors and filter them out of the input data.
Default: False
Output options
- --stdfreq
Frequency to which the physiological signals are resampled for output. Default is 25.
Default: 25.0
Phase projection tuning
- --outputbins
Number of output phase bins (default is 32).
Default: 32
- --gridbins
Width of the gridding kernel in output phase bins (default is 3.0).
Default: 3.0
- --gridkernel
Possible choices: old, gauss, kaiser
Convolution gridding kernel. Default is kaiser
Default: “kaiser”
- --projmask
Phase projection will be restricted to voxels in MASKNAME (overrides normal intensity mask.)
- --projectwithraw
Use fMRI derived cardiac waveform as phase source for projection, even if a plethysmogram is supplied.
Default: False
- --fliparteries
Attempt to detect arterial signals and flip over the timecourses after phase projection (since relative arterial blood susceptibility is inverted relative to venous blood).
Default: False
- --arteriesonly
Restrict cardiac waveform estimation to putative arteries only.
Default: False
Version options
- --version
show program’s version number and exit
- --detailedversion
show program’s version number and exit
Miscellaneous options.
- --aliasedcorrelation
Attempt to calculate absolute delay using an aliased correlation (experimental).
Default: False
- --upsample
Attempt to temporally upsample the fMRI data (experimental).
Default: False
- --estimateflow
Estimate blood flow using optical flow (experimental).
Default: False
- --noprogressbar
Will disable showing progress bars (helpful if stdout is going to a file).
Default: True
- --infotag
Additional key, value pairs to add to the info json file (useful for tracking analyses).
Debugging options (probably not of interest to users)
- --debug
Turn on debugging information.
Default: False
- --nodetrend
Disable data detrending.
Default: 3
- --noorthog
Disable orthogonalization of motion confound regressors.
Default: True
- --disablenotch
Disable subharmonic notch filter.
Default: False
- --nomask
Disable data masking for calculating cardiac waveform.
Default: True
- --nocensor
Bad points will not be excluded from analytic phase projection.
Default: True
- --noappsmooth
Disable smoothing app file in the phase direction.
Default: True
- --nophasefilt
Disable the phase trend filter (probably not a good idea).
Default: True
- --nocardiacalign
Disable alignment of pleth signal to fMRI derived cardiac signal.
Default: True
- --saveinfoastext
Save the info file in text format rather than json.
Default: True
- --saveintermediate
Save some data from intermediate passes to help debugging.
Default: False
- --increaseoutputlevel
Increase the number of intermediate output files.
Default: 0
- --decreaseoutputlevel
Decrease the number of intermediate output files.
Default: 0
Example:
Extract the cardiac waveform and generate phase projections
Case 1: When you don’t have a pleth recording
There are substantial improvements to the latest versions of happy. In the old versions, you actually had to run happy twice - the first time to estimate the vessel locations, and the second to actually derive the waveform. Happy now combines these operations interpolation a single run with multiple passes - the first pass locates voxels with high variance, labels them as vessels, then reruns the derivation, restricting the cardiac estimation to these high variance voxels. This gives substantially better results.
Using the example data in the example directory, try the following:
happy \ rapidtide/data/examples/src/sub-HAPPYTEST.nii.gz \ rapidtide/data/examples/src/sub-HAPPYTEST.json \ rapidtide/data/examples/dst/happytest
This will perform a happy analysis on the example dataset. To see the extracted cardiac waveform (original and filtered), you can use showtc (also part of them rapidtide package):
showtc \ rapidtide/data/examples/src/happytest_desc-slicerescardfromfmri_timeseries.json:cardiacfromfmri,cardiacfromfmri_dlfiltered \ --format separate
rapidtide2std
Description:
This is a utility for registering rapidtide output maps to standard coordinates. It’s usually much faster to run rapidtide in native space then transform afterwards to MNI152 space. NB: this will only work if you have a working FSL installation.
Inputs:
Outputs:
New versions of the rapidtide output maps, registered to either MNI152 space or to the hires anatomic images for the subject. All maps are named with the specified root name with ‘_std’ appended.
Usage:
usage: rapidtide2std INPUTFILEROOT OUTPUTDIR FEATDIRECTORY [--all] [--hires] required arguments: INPUTFILEROOT - The base name of the rapidtide maps up to but not including the underscore OUTPUTDIR - The location for the output files FEADDIRECTORY - A feat directory (x.feat) where registration to standard space has been performed optional arguments: --all - also transform the corrout file (warning - file may be huge) --hires - transform to match the high resolution anatomic image rather than the standard --linear - only do linear transformation, even if warpfile exists
showxcorr_legacy
Description:
Like rapidtide, but for single time courses. Takes two text files as input, calculates and displays the time lagged crosscorrelation between them, fits the maximum time lag, and estimates the significance of the correlation. It has a range of filtering, windowing, and correlation options. This is the old interface - for new analyses you should use showxcorrx.
Inputs:
showxcorr requires two text files containing timecourses with the same sample rate, one timepoint per line, which are to be correlated, and the sample rate.
Outputs:
showxcorr outputs everything to standard out, including the Pearson correlation, the maximum cross correlation, the time of maximum cross correlation, and estimates of the significance levels (if specified). There are no output files.
Usage:
usage: showxcorr timecourse1 timecourse2 samplerate [-l LABEL] [-s STARTTIME] [-D DURATION] [-d] [-F LOWERFREQ,UPPERFREQ[,LOWERSTOP,UPPERSTOP]] [-V] [-L] [-R] [-C] [-t] [-w] [-f] [-z FILENAME] [-N TRIALS] required arguments: timcoursefile1: text file containing a timeseries timcoursefile2: text file containing a timeseries samplerate: the sample rate of the timecourses, in Hz optional arguments: -t - detrend the data -w - prewindow the data -l LABEL - label for the delay value -s STARTTIME - time of first datapoint to use in seconds in the first file -D DURATION - amount of data to use in seconds -r RANGE - restrict peak search range to +/- RANGE seconds (default is +/-15) -d - turns off display of graph -F - filter data and regressors from LOWERFREQ to UPPERFREQ. LOWERSTOP and UPPERSTOP can be specified, or will be calculated automatically -V - filter data and regressors to VLF band -L - filter data and regressors to LFO band -R - filter data and regressors to respiratory band -C - filter data and regressors to cardiac band -T - trim data to match -A - print data on a single summary line -a - if summary mode is on, add a header line showing what values mean -f - negate (flip) second regressor -z FILENAME - use the columns of FILENAME as controlling variables and return the partial correlation -N TRIALS - estimate significance thresholds by Monte Carlo with TRIALS repetition
showxcorrx
Description:
This is the newest, most avant-garde version of showxcorr. Because it’s an x file, it’s more fluid and I don’t guarantee that it will keep a stable interface (or even work at any given time). But every time I add something new, it goes here. The goal is eventually to make this the “real” version. Unlike rapidtide, however, I’ve let it drift quite a bit without syncing it because some people here actually use showxcorr and I don’t want to disrupt workflows…
Inputs:
showxcorrx requires two text files containing timecourses with the same sample rate, one timepoint per line, which are to be correlated, and the sample rate.
Outputs:
showxcorrx outputs everything to standard out, including the Pearson correlation, the maximum cross correlation, the time of maximum cross correlation, and estimates of the significance levels (if specified). There are no output files.
Usage:
showtc
Description:
A very simple command line utility that takes a text file and plots the data in it in a matplotlib window. That’s it. A good tool for quickly seeing what’s in a file. Has some options to make the plot prettier.
Inputs:
Text files containing time series data
Outputs:
None
Usage:
Plots the data in text files.
usage: showtc [-h] [--samplerate FREQ | --sampletime TSTEP]
[--displaytype {time,power,phase}]
[--format {overlaid,separate,separatelinked}] [--waterfall]
[--voffset OFFSET] [--transpose] [--normall] [--title TITLE]
[--xlabel LABEL] [--ylabel LABEL]
[--legends LEGEND[,LEGEND[,LEGEND...]]] [--legendloc LOC]
[--colors COLOR[,COLOR[,COLOR...]]] [--nolegend] [--noxax]
[--noyax] [--linewidth LINEWIDTH[,LINEWIDTH[,LINEWIDTH...]]]
[--tofile FILENAME] [--fontscalefac FAC] [--saveres DPI]
[--starttime START] [--endtime END] [--numskip NUM] [--debug]
[--version] [--detailedversion]
textfilenames [textfilenames ...]
Positional Arguments
- textfilenames
One or more input files, with optional column specifications
Named Arguments
- --samplerate
Set the sample rate of the data file to FREQ. If neither samplerate or sampletime is specified, sample rate is 1.0.
Default: auto
- --sampletime
Set the sample rate of the data file to 1.0/TSTEP. If neither samplerate or sampletime is specified, sample rate is 1.0.
Default: auto
- --displaytype
Possible choices: time, power, phase
Display data as time series (default), power spectrum, or phase spectrum.
Default: “time”
- --format
Possible choices: overlaid, separate, separatelinked
Display data overlaid (default), in individually scaled windows, or in separate windows with linked scaling.
Default: “overlaid”
- --waterfall
Display multiple timecourses in a waterfall plot.
Default: False
- --voffset
Plot multiple timecourses with OFFSET between them (use negative OFFSET to set automatically).
Default: 0.0
- --transpose
Swap rows and columns in the input files.
Default: False
- --normall
Normalize all displayed timecourses to unit standard deviation and zero mean.
Default: False
- --starttime
Start plotting at START seconds (default is the start of the data).
- --endtime
Finish plotting at END seconds (default is the end of the data).
- --numskip
Skip NUM lines at the beginning of each file (to get past header lines).
Default: 0
- --debug
Output additional debugging information.
Default: False
General plot appearance options
- --title
Use TITLE as the overall title of the graph.
Default: “”
- --xlabel
Label for the plot x axis.
Default: “”
- --ylabel
Label for the plot y axis.
Default: “”
- --legends
Comma separated list of legends for each timecourse.
- --legendloc
Integer from 0 to 10 inclusive specifying legend location. Legal values are: 0: best, 1: upper right, 2: upper left, 3: lower left, 4: lower right, 5: right, 6: center left, 7: center right, 8: lower center, 9: upper center, 10: center. Default is 2.
Default: 2
- --colors
Comma separated list of colors for each timecourse.
- --nolegend
Turn off legend label.
Default: True
- --noxax
Do not show x axis.
Default: True
- --noyax
Do not show y axis.
Default: True
- --linewidth
A comma separated list of linewidths (in points) for plots. Default is 1.
- --tofile
Write figure to file FILENAME instead of displaying on the screen.
- --fontscalefac
Scaling factor for annotation fonts (default is 1.0).
Default: 1.0
- --saveres
Write figure to file at DPI dots per inch (default is 1000).
Default: 1000
Version options
- --version
show program’s version number and exit
- --detailedversion
show program’s version number and exit
glmfilt
Description:
Uses a GLM filter to remove timecourses (1D text files or 4D NIFTI files) from 4D NIFTI files.
Inputs:
Outputs:
Usage:
Fits and removes the effect of voxel specific and/or global regressors.
usage: glmfilt [-h] [--numskip NUMSKIP] [--evfile EVFILE [EVFILE ...]]
[--dmask DATAMASK] [--limitoutput]
inputfile outputroot
Positional Arguments
- inputfile
The name of the 3 or 4 dimensional nifti file to fit.
- outputroot
The root name for all output files.
Named Arguments
- --numskip
The number of points to skip at the beginning of the timecourse when fitting. Default is 0.
Default: 0
- --evfile
One or more files (text timecourse or 4D NIFTI) containing signals to regress out.
- --dmask
Use DATAMASK to specify which voxels in the data to use.
- --limitoutput
Only save the filtered data and the R value.
Default: True
temporaldecomp
Description:
Inputs:
Outputs:
Usage:
Perform PCA or ICA decomposition on a data file in the time dimension.
usage: temporaldecomp [-h] [--dmask DATAMASK] [--ncomp NCOMPS]
[--smooth SIGMA] [--type {pca,sparse,ica}] [--nodemean]
[--novarnorm]
datafile outputroot
Positional Arguments
- datafile
The name of the 3 or 4 dimensional nifti file to fit
- outputroot
The root name for the output nifti files
Named Arguments
- --dmask
Use DATAMASK to specify which voxels in the data to use.
- --ncomp
The number of PCA/ICA components to return (default is to estimate the number).
Default: -1.0
- --smooth
Spatially smooth the input data with a SIGMA mm kernel.
Default: 0.0
- --type
Possible choices: pca, sparse, ica
Type of decomposition to perform. Default is pca.
Default: “pca”
- --nodemean
Do not demean data prior to decomposition.
Default: True
- --novarnorm
Do not variance normalize data prior to decomposition.
Default: True
spatialdecomp
Description:
Inputs:
Outputs:
Usage:
Perform PCA or ICA decomposition on a data file in the spatial dimension.
usage: spatialdecomp [-h] [--dmask DATAMASK] [--ncomp NCOMPS] [--smooth SIGMA]
[--type {pca,sparse,ica}] [--nodemean] [--novarnorm]
datafile outputroot
Positional Arguments
- datafile
The name of the 3 or 4 dimensional nifti file to fit
- outputroot
The root name for the output nifti files
Named Arguments
- --dmask
Use DATAMASK to specify which voxels in the data to use.
- --ncomp
The number of PCA/ICA components to return (default is to estimate the number).
Default: -1.0
- --smooth
Spatially smooth the input data with a SIGMA mm kernel.
Default: 0.0
- --type
Possible choices: pca, sparse, ica
Type of decomposition to perform. Default is pca.
Default: “pca”
- --nodemean
Do not demean data prior to decomposition.
Default: True
- --novarnorm
Do not variance normalize data prior to decomposition.
Default: True
polyfitim
Description:
Inputs:
Outputs:
Usage:
Fit a spatial template to a 3D or 4D NIFTI file.
usage: polyfitim [-h] [--regionatlas ATLASFILE] [--order ORDER]
datafile datamask templatefile templatemask outputroot
Positional Arguments
- datafile
The name of the 3 or 4 dimensional nifti file to fit.
- datamask
The name of the 3 or 4 dimensional nifti file valid voxel mask (must match datafile).
- templatefile
The name of the 3D nifti template file (must match datafile).
- templatemask
The name of the 3D nifti template mask (must match datafile).
- outputroot
The root name for all output files.
Named Arguments
- --regionatlas
Do individual fits to every region in ATLASFILE (3D NIFTI file).
- --order
Perform fit to ORDERth order (default (and minimum) is 1)
Default: 1
histnifti
Description:
A command line tool to generate a histogram for a nifti file
Inputs:
A nifti file
Outputs:
A text file containing the histogram information
None
Usage:
Generates a histogram of the values in a NIFTI file.
usage: histnifti [-h] [--histlen LEN] [--minval MINVAL] [--maxval MAXVAL]
[--robustrange] [--transform] [--nozero]
[--nozerothresh THRESH] [--normhist] [--maskfile MASK]
[--nodisplay]
inputfile outputroot
Positional Arguments
- inputfile
The name of the input NIFTI file.
- outputroot
The root of the output file names.
Named Arguments
- --histlen
Set histogram length to LEN (default is to set automatically).
- --minval
Minimum bin value in histogram.
- --maxval
Maximum bin value in histogram.
- --robustrange
Set histogram limits to the data’s robust range (2nd to 98th percentile).
Default: False
- --transform
Replace data value with it’s percentile score.
Default: False
- --nozero
Do not include zero values in the histogram.
Default: False
- --nozerothresh
Absolute values less than this are considered zero. Default is 0.01.
Default: 0.01
- --normhist
Return a probability density instead of raw counts.
Default: False
- --maskfile
Only process voxels within the 3D mask MASK.
- --nodisplay
Do not display histogram.
Default: True
showhist
Description:
Another simple command line utility that displays the histograms generated by rapidtide.
Inputs:
A textfile generated by rapidtide containing histogram information
Outputs:
None
Usage:
usage: showhist textfilename plots xy histogram data in text file required arguments: textfilename - a text file containing one timepoint per line
resamp1tc
Description:
This takes an input text file at some sample rate and outputs a text file resampled to the specified sample rate.
Inputs:
Outputs:
Usage:
resamp1tc - resample a timeseries file usage: resamp1tc infilename insamplerate outputfile outsamplerate [-s] required arguments: inputfile - the name of the input text file insamplerate - the sample rate of the input file in Hz outputfile - the name of the output text file outsamplerate - the sample rate of the output file in Hz options: -s - split output data into physiological bands (LFO, respiratory, cardiac)
resamplenifti
Description:
This takes an input nifti file at some TR and outputs a nifti file resampled to the specified TR.
Inputs:
Outputs:
Usage:
usage: resamplenifti inputfile inputtr outputname outputtr [-a] required arguments: inputfile - the name of the input nifti file inputtr - the tr of the input file in seconds outputfile - the name of the output nifti file outputtr - the tr of the output file in seconds options: -a - disable antialiasing filter (only relevant if you are downsampling in time)
tcfrom3col
Description:
A simple command line that takes an FSL style 3 column regressor file and generates a time course (waveform) file. FSL 3 column files are text files containing one row per “event”. Each row has three columns: start time in seconds, duration in seconds, and waveform value. The output waveform is zero everywhere that is not covered by an “event” in the file.
Inputs:
A three column text file
Outputs:
A single column text file containing the waveform
Usage:
tcfrom3col - convert a 3 column fsl style regressor into a one column timecourse usage: tcfrom3col infile timestep numpoints outfile required arguments: infile: a text file containing triplets of start time, duration, and value timestep: the time step of the output time coures in seconds numpoints: the number of output time points outfile: the name of the output time course file
pixelcomp
Description:
A program to compare voxel values in two 3D NIFTI files. You give pixelcomp two files, each with their own mask. Any voxel that has a nonzero mask in both files gets added to a list of xy pairs, with the value from the first file being x, and the value from the second file being y. Pixelcomp then: 1) Makes and displays a 2D histogram of all the xy values. 2) Does a linear fit to x and y, and outputs the coefficients (slope and offset) to a XXX_linfit.txt file. 3) Writes all the xy pairs to a tab separated text file, and 4) Makes a Bland-Altman plot of x vs y
Inputs:
Two 3D NIFTI image files, the accompanying mask files, and the root name for the output files.
Outputs:
None
Usage:
showtc - plots the data in text files usage: showtc texfilename[:col1,col2...,coln] [textfilename]... [--nolegend] [--pspec] [--phase] [--samplerate=Fs] [--sampletime=Ts] required arguments: textfilename - a text file containing whitespace separated timecourses, one timepoint per line A list of comma separated numbers following the filename and preceded by a colon is used to select columns to plot optional arguments: --nolegend - turn off legend label --pspec - show the power spectra magnitudes of the input data instead of the timecourses --phase - show the power spectra phases of the input data instead of the timecourses --transpose - swap rows and columns in the input files --waterfall - plot multiple timecourses as a waterfall --voffset=VOFFSET - plot multiple timecourses as with VOFFSET between them (use negative VOFFSET to set automatically) --samplerate=Fs - the sample rate of the input data is Fs Hz (default is 1Hz) --sampletime=Ts - the sample time (1/samplerate) of the input data is Ts seconds (default is 1s) --colorlist=C1,C2,.. - cycle through the list of colors specified by CN --linewidth=LW - set linewidth to LW points (default is 1) --fontscalefac=FAC - scale all font sizes by FAC (default is 1.0) --legendlist=L1,L2,.. - cycle through the list of legends specified by LN --tofile=FILENAME - write figure to file FILENAME instead of displaying on the screen --title=TITLE - use TITLE as the overall title of the graph --separate - use a separate subplot for each timecourse --separatelinked - use a separate subplot for each timecourse, but use a common y scaling --noxax - don't show x axis --noyax - don't show y axis --starttime=START - start plot at START seconds --endtime=END - end plot at END seconds --legendloc=LOC - Integer from 0 to 10 inclusive specifying legend location. Legal values are: 0: best, 1: upper right, 2: upper left, 3: lower left, 4: lower right, 5: right, 6: center left, 7: center right, 8: lower center, 9: upper center, 10: center. Default is 2. --debug - print debugging information
glmfilt
Description:
Uses a GLM filter to remove timecourses (1D text files or 4D NIFTI files) from 4D NIFTI files.
Inputs:
Outputs:
Usage:
usage: glmfilt datafile numskip outputroot evfile [evfile_2...evfile_n] Fits and removes the effect of voxel specific and/or global regressors
ccorrica
Description:
Find temporal crosscorrelations between all the columns in a text file (for example the timecourse files output by MELODIC.)
Inputs:
Outputs:
Usage:
ccorrica - find temporal crosscorrelations between ICA components usage: ccorrica timecoursefile TR timcoursefile: text file containing multiple timeseries, one per column, whitespace separated TR: the sample period of the timecourse, in seconds
showstxcorr
Description:
Calculate and display the short term crosscorrelation between two timeseries (useful for dynamic correlation).
Inputs:
Outputs:
Usage:
showstxcorr - calculate and display the short term crosscorrelation between two timeseries usage: showstxcorr -i timecoursefile1 [-i timecoursefile2] --samplefreq=FREQ -o outputfile [-l LABEL] [-s STARTTIME] [-D DURATION] [-d] [-F LOWERFREQ,UPPERFREQ[,LOWERSTOP,UPPERSTOP]] [-V] [-L] [-R] [-C] [--nodetrend] [-nowindow] [-f] [--phat] [--liang] [--eckart] [-z FILENAME] required arguments: -i, --infile= timcoursefile1 - text file containing one or more timeseries [-i, --infile= timcoursefile2] - text file containing a timeseries NB: if one timecourse file is specified, each column is considered a timecourse, and there must be at least 2 columns in the file. If two filenames are given, each file must have only one column of data. -o, --outfile=OUTNAME: - the root name of the output files --samplefreq=FREQ - sample frequency of all timecourses is FREQ or --sampletime=TSTEP - time step of all timecourses is TSTEP NB: --samplefreq and --sampletime are two ways to specify the same thing. optional arguments: --nodetrend - do not detrend the data before correlation --nowindow - do not prewindow data before corrlation --phat - perform phase alignment transform (PHAT) rather than standard crosscorrelation --liang - perform phase alignment transform with Liang weighting function rather than standard crosscorrelation --eckart - perform phase alignment transform with Eckart weighting function rather than standard crosscorrelation -s STARTTIME - time of first datapoint to use in seconds in the first file -D DURATION - amount of data to use in seconds -d - turns off display of graph -F - filter data and regressors from LOWERFREQ to UPPERFREQ. LOWERSTOP and UPPERSTOP can be specified, or will be calculated automatically -V - filter data and regressors to VLF band -L - filter data and regressors to LFO band -R - filter data and regressors to respiratory band -C - filter data and regressors to cardiac band -W WINDOWLEN - use a window length of WINDOWLEN seconds (default is 50.0s) -S STEPSIZE - timestep between subsequent measurements (default is 25.0s). Will be rounded to the nearest sample time -f - negate second regressor
tidepool
Description:
Tidepool is a handy tool for displaying all of the various maps generated by rapidtide in one place, overlayed on an anatomic image. This makes it easier to see how all the maps are related to one another. To use it, launch tidepool from the command line, navigate to a rapidtide output directory, and then select a lag time (maxcorr) map. tidpool will figure out the root name and pull in all of the other associated maps, timecourses, and info files. The displays are live, and linked together, so you can explore multiple parameters efficiently. Works in native or standard space.
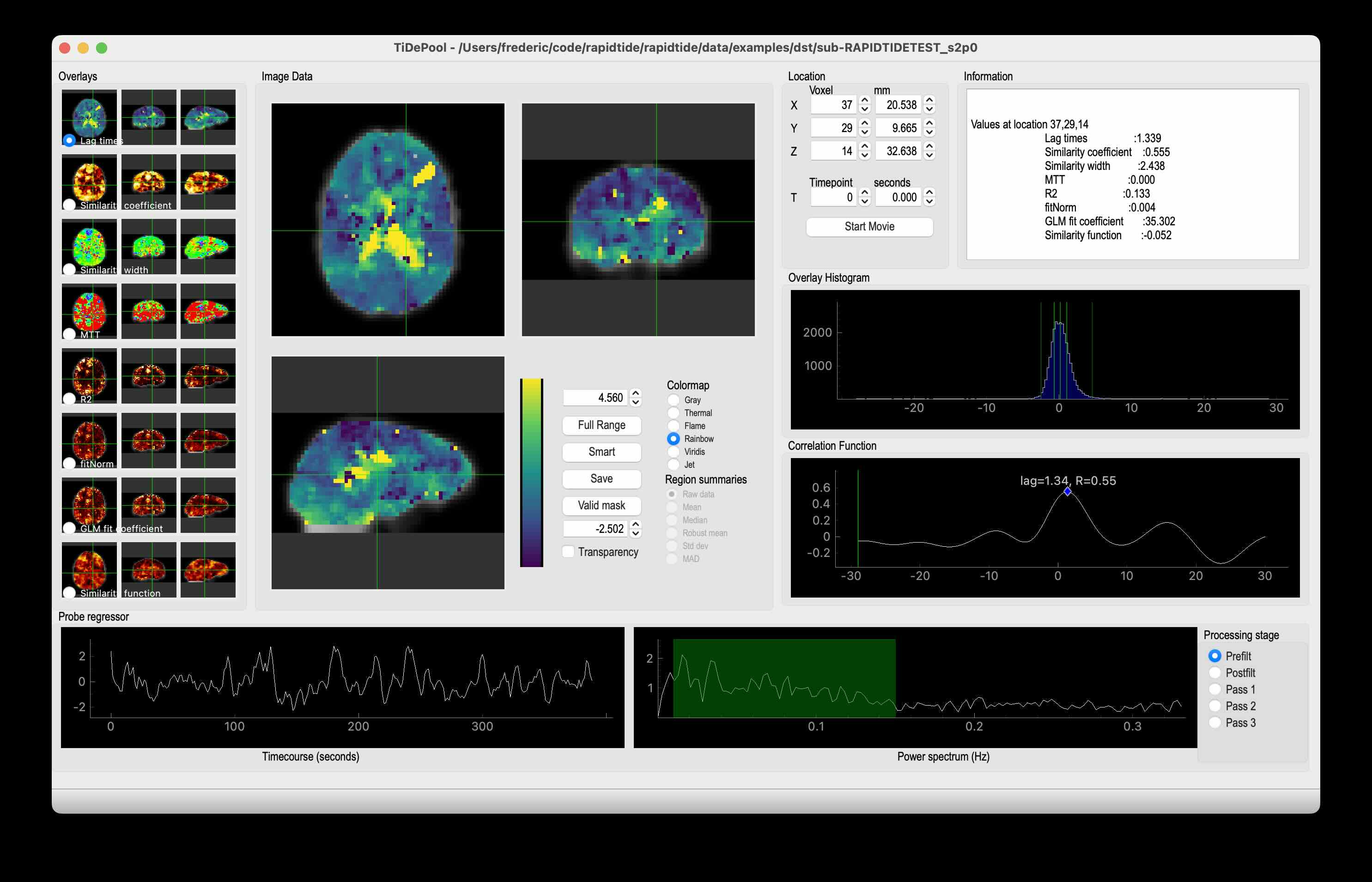
The main tidepool window with a dataset loaded.
Inputs:
Tidepool loads most of the output files from a rapidtide analysis. The files must all be in the same directory, and use the naming convention and file formats that rapidtide uses.
Features:
There are many panels to the tidepool window. They are described in detail below.
Image Data
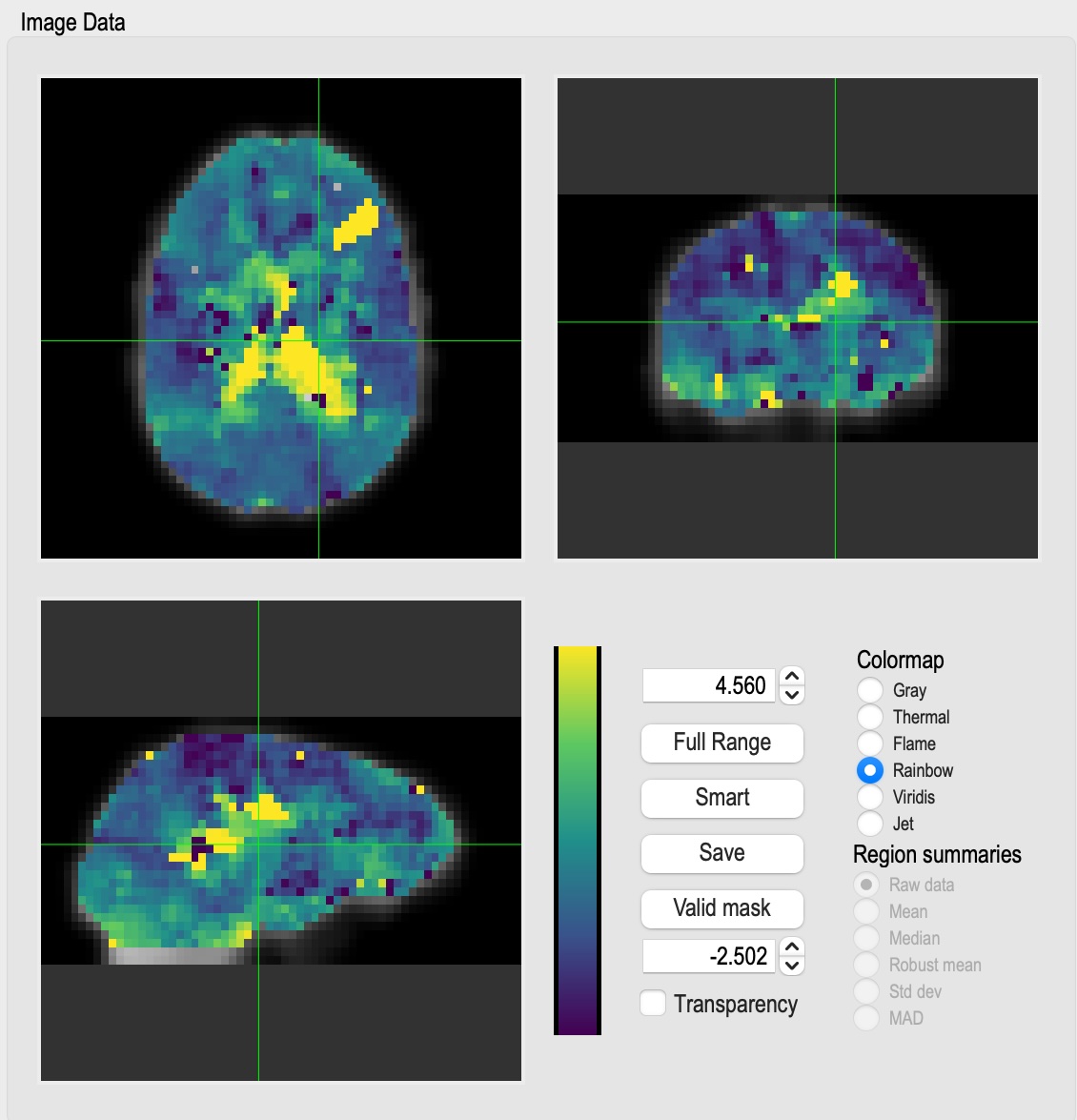
This is the main control of the tidepool window. This shows three orthogonal views of the active map (maxtime in this case) superimposed on an anatomic image (the mean fmri input image to rapidtide by default). Use the left mouse button to select a location in any of the images, and the other two will update to match. The intersecting green lines show the lower left corner of the active location. The lower righthand panel allows you to adjust various parameters, such as the minimum and maximum values of the colormap (set to the “robust range” by default). The “Transparency” button toggles whether values outside of the active range are set to the minimum or maximum colormap value, or are not displayed. The radio buttons in the upper right section of the colormap control panel allow you to change to colormap used from the default values. The “Full Range” button sets the colormap limits to the minimum and maximum values in the map. The “Smart” button sets the colormap limits to the 2% to 98% limits (the “robust range”). The “Save” button saves the three active images to jpeg files. The mask button (below the “Smart” button) indicates what mask to apply when displaying the map. By default, this is the “Valid” mask - all voxels where the rapidtide fit converged. Right clicking on this button gives you a popup window which allows you to select from several other masks, including no mask, the voxels used to set the initial regressor, the voxels used in the final refinement pass, and a range of significance values for the rapidtide fit.
The popup menu for selecting the display mask.
Overlay Selector
This panel allows you to select which map is displayed in the “Image Data” panel using the radio buttons in the corner of each image. The maps displayed will vary based on the analysis performed. These are all three dimensional maps, with the exception of the bottom map shown - the “Similarity function”. This is the full correlation (or other similarity function) used by rapidtide to generate the various maps. When this is loaded, you can use the controls in the “Location” panel to select different time points, or to show the function as a movie.
Information panel

This panel shows the location of the cursor in the “Image Data” panel, and the value of all the loaded maps at that location. If the rapidtide fit failed at that location, all values will be set to zero, and there will be a text description of the reason for the fit failure.
Histogram
This panel shows the histogram of values displayed (i.e. those selected by the current active mask) in the “Image Data” panel. By default the range shown is the search range specified during the rapidtide analysis. You can pand and zoom the histogram by clicking and holding the left or right mouse button and moving the mouse. The green bars on the graph show the 2%, 25%, 50%, 75%, and 98% percentile values of the histogram.
This shows the result of zooming the histogram using the right mouse button. With the mouse in the panel, left click on the “A” in the square box in the lower left of the plot to restore the default display values.
Similarity Function
This panel shows the similarity function (correlation, mutual information) at the location of the cursor in the “Image Data” window. There is a marker showing the maxtime and maxcorr found by the fit (or the text “No valid fit” if the fit failed). This can be used for diagnosing strange fit behavior.
Probe Regressor
This panel shows the probe regressor used in various stages of the rapidtide analysis. The left panel shows the time domain, the right shows the frequency domain, with a translucent green overlay indicating the filter band used in the analysis. The radio buttons on the right select which analysis stage to display: “Prefilt” is the initial probe regressor, either the global mean, or an externally supplied timecourse; “Postfilt” is this regressor after filtering to the active analysis band. “PassX” is the resampled regressor used in each of the analysis passes.
Usage:
If tidepool is called without arguments, a dialog box will appear to allow you to select the maxtime map from the dataset you want to load. This (and other things) can alternately be supplied on the command line as specified below.